The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know: Part 2
 |
|
|
|

This is part 2 of a two-part blog series covering our whitepaper, “The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know” written by Vincent Balgos, Medial Solution Manager at Jama Software. In this paper, Vincent discusses the In Vitro Diagnostic Regulation (IVDR), developed by the EU Commission (CE), which was created to replace the previous In Vitro Diagnostic Directive (IVDD).
Part 1 of this blog series is available HERE. To read the whitepaper in its entirety, download it HERE.
With more than 150 pages of regulations, there were many changes to strengthen and grow the path to IVDs marketed and distributed in the EU. The IVDR provides a more comprehensive approach to regulating devices as it encompasses the entire product lifecycle: from initial concept to design and manufacturing, to continual on-market support along maintaining good documentation practices. For the purpose of this paper,
only a few selected topics will be covered in detail with some additional insights from an industry perspective.
Who is impacted?
Medical companies that develop IVD’s marketed to the European Union, and its population. This includes non-EU-based companies that have products in the EU region.
What is impacted?
In-Vitro Diagnostics (IVDs) products and accessories that are used to perform tests on various sample types to help diagnose a condition, detect infections, or monitor drug responses.
When is it happening?
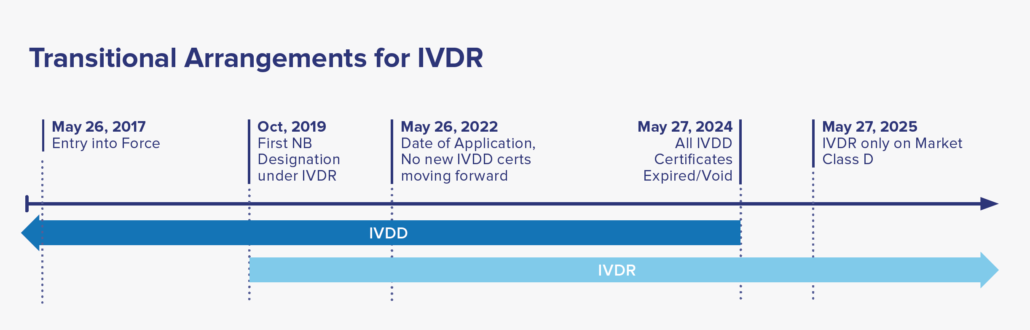
Date of Application: May 26, 2022, where only IVDR applications will be accepted by NB. A two-year window period afterwards to allow companies to transition their IVDD to IVDR certification. By May 2025, all IVDD certificates will be voided, with IVDR covering all placed IVDs in market.
What countries are impacted?
European Union and the United Kingdom specifically, but companies from around the world who have products in the UK or EU are also required to conform.
Why are things changing?
To improve the safety and quality of IVDs in the EU market.
A key significant change of the new regulation is how IVD’s are classified. Similar to the previous directive, a risk-based approach (with respects to public and patient) is used to classify IVDs under the new IVDR framework. There are four main classes as listed in the table below, established by seven classifications rules defined in Annex VIII of IVDR.
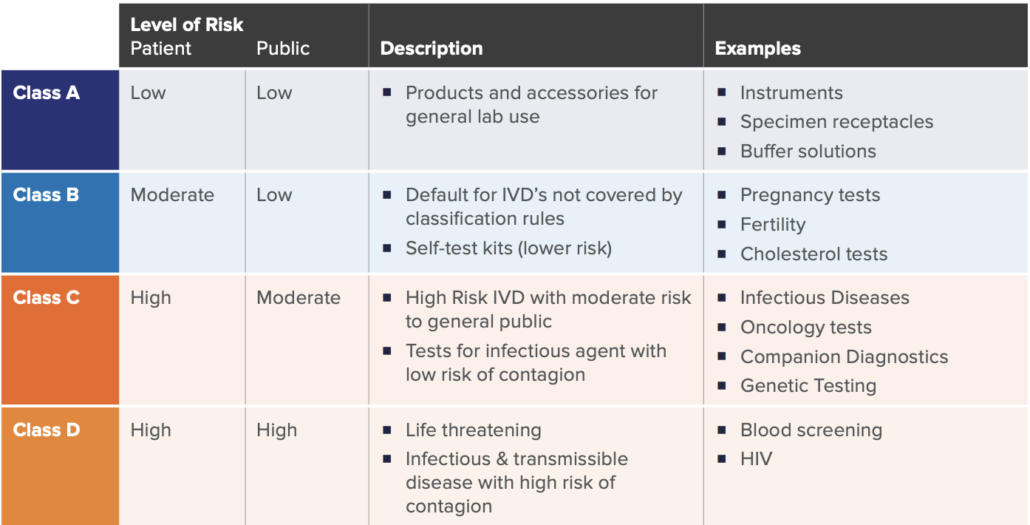
While there may be nuances to the rules set, these four categories broadly cover the majority of the IVD spectrum. This new classification schema only allows Class A devices to be self-certified by manufacturers, whereas Class B, C, and D require more assessment and certification by notified bodies.
As in similar regulatory pathways, device classification is significant in determining the overall requirements, as the higher the IVD risks, the more onerous the regulatory requirements, and the higher the involvement with an external Notified Body. For example, a new HIV Test would be categorized as Class D, which would require the highest number of internal activities during design, development, on-market support, and associated documentation. This would also require the highest amount of interaction, assessment, and certification from the Notified Body. Furthermore, specific regulations such Post-Market Surveillance, Quality Management System elements, and annual updates to reports are required for higher risk class (C and D) devices.
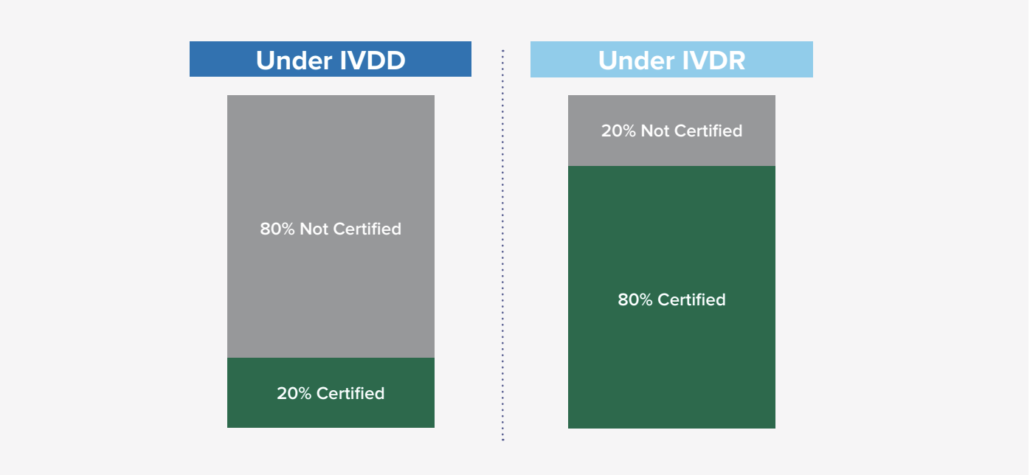
Based on general research from industry subject matter experts, it was estimated that only 20% required Notified Body certification under the previous IVDD, while 80% did not require certification. With the new classification schema and requirements under IVDR, that ratio has flipped where it is expected that the majority of IVD’s (80%) will now need some Notified Body involvement. This new shift (in engaging Notified Bodies and the new requirement) is significant in many ways as it not only impacts the manufacturers, but also the Notified Bodies as demand for their engagement has risen exponentially. There are some concerns about the current Notified Body capacity, so it is encouraged to start engaging with a Notified Body proactively, as the backlog to engage could be longer than anticipated.
For certified IVD’s that are currently on-market classified under the IVDD guidance, reclassification to the IVDR categories and recertification to meet the IVDR is required for continual sale and distribution to EU market. Under the IVDR, there are no grandfathering clauses to allow the IVDD devices to remain on market after May 2025. Considering there are many IVD’s in use, the EU established a five-year timeline to allow manufacturers to transition to IVDR. See the timeline below for more details.
 Since IVDR’s announcement in 2017, many companies and SME’s (including this author) started to update
Since IVDR’s announcement in 2017, many companies and SME’s (including this author) started to update
their internal procedures, adjust development and documentation activities, and hire additional resources
in response to the impending changes. In addition, remediations to current devices’ Design History Files
(DHFs) to align with regulations were also underway. These include adding additional testing for new
requirements such as performance studies, clinical evaluations, etc. These activities may be significant,
and a major resource pull from other ongoing projects. Therefore, it’s critical to acknowledge that the new
IVDR regulations impact not only future but current IVDs on the market as well.
Under Article 15, a new IVDR requirement is that manufacturers are required to have a regulatory compliance expert in their organization to be responsible for the compliance of the in-vitro diagnostics regulations. This person must be a qualified regulatory expert with previous demonstrated qualification such as 1) formal certification from approved regulatory body and/or 2) minimum of four years of industry experience as a regulatory affairs professional in the IVD field. This role (new for some organizations) provides general regulatory affairs guidance, interpretation of regulations to internal teams, and helps facilitate discussions with Notified Bodies, regulatory agencies, and EU Competent Authorities.
While not a new requirement to IVD practices, Annex I Chapter I of the IVDR has multiple languages referring to and establishing risk management practices. This further substantiates the EU focus on a riskbased approach when developing devices and encourages many best practices that Jama Software® has seen many of our IVD customers follow.
This new language includes the following requirements:
This updated language continues the industry practice of risk management that is further established in ISO 14971 “Medical Devices – Application of Risk management to Medical Devices” and TR 24971 “Medical Devices – Guidance on the application of ISO 14971.” Based on the reasons why the IVDR came into fruition (PIP accidents), it can be surmised that an organization’s risk management process will be under significant scrutiny by the Notified Body. Therefore, Risk Management Procedures have been a focal point of update for organizations to strengthen risk practices and ensure compliance. Remediation of risk files may also be warranted for devices currently on the market, or soon to be on the market in the EU.
Based on this author’s experience, this risk activity alone requires significant time and resources to accomplish. Considering some risk files could have significate number of documents (plans, evaluations, reports) with details that require comprehensive review from many stakeholders, this is an effort that needs formal organization support to successfully comply with the IVDR and its compliance timeline. Therefore, it is recommended to prioritize
appropriately and revisit the Risk Management section, and other impacted areas of the IVDR as soon as possible.
Based on discussions with various IVD customers, general research, and internal experience, we recommend the following guidance:
As with many types of changes in regulations, these have substantial impact on how organizations and their teams operate in the design, development, and manufacturing documentation of IVDs. It is encouraged to proactively review these new regulations as it may require significant time and resource to adapt to continue developing IVD’s for the European market.
DISCLAIMER
Jama Software is not an accredited regulatory subject matter expert, so these are general guidance and insights from working with many IVD customers, general research, and some internal experience. It is suggested to work with a certified Regulatory Affairs consultant for formal recommendations for your organization.
References:
1. https://www.bsigroup.com/meddev/LocalFiles/en-IN/Technologies/BSI-md-ivd-diagnostic-directive-guide-brochure-UK-EN.pdf
2. https://ec.europa.eu/growth/single-market/ce-marking_en
With the new IVDR, it is expected that manufacturers will need to shift to a more regimented process of developing, manufacturing, and managing IVD’s. Similar to other regulatory pathways, good requirements management is the best practice in ensuring compliance with regulations, reducing risk, and launching safe and effective products.
Jama Connect® for Medical Device Development helps medical device teams reduce the effort required to achieve regulatory compliance throughout the development process. With this solution, medical device teams can manage design controls for device requirements and related risks, simplifying regulatory submissions and audit preparations while accelerating time to market. Jama Connect creates a digital thread for systems engineering and
ensures Live Traceability™ and alignment across the product development lifecycle to seamlessly connect development solutions and facilitate product success.
ABOUT THE AUTHOR, VINCENT BALGOS
Vincent Balgos currently leads the Medical Solution at Jama Software. Prior to joining Jama Software, he worked in the medical device / IVD industry for over 17 years with roles in systems engineering, product development and project management. Vincent has successful history in launching new products to the global regulated market, and is experienced in product development, risk management, quality systems, and medical device regulations.
Part 1 of this blog series is available HERE. To read the whitepaper in its entirety, download it HERE.