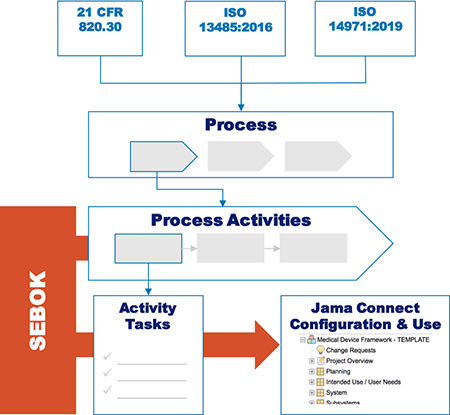
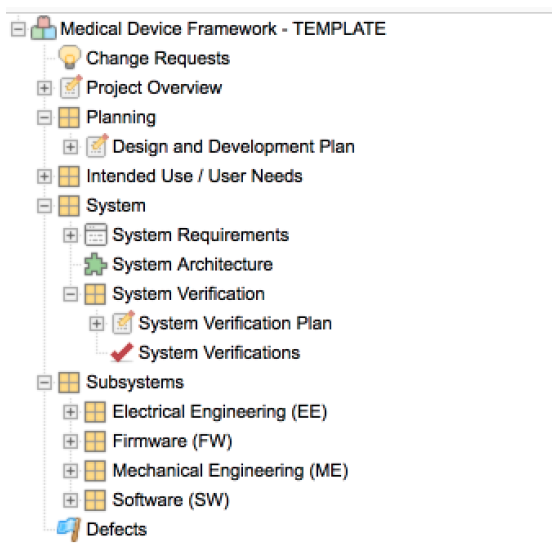
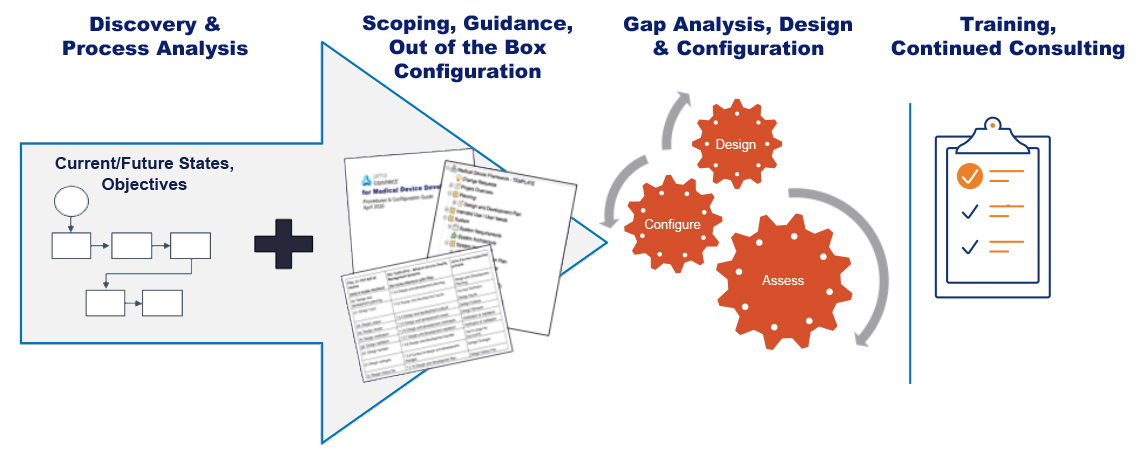
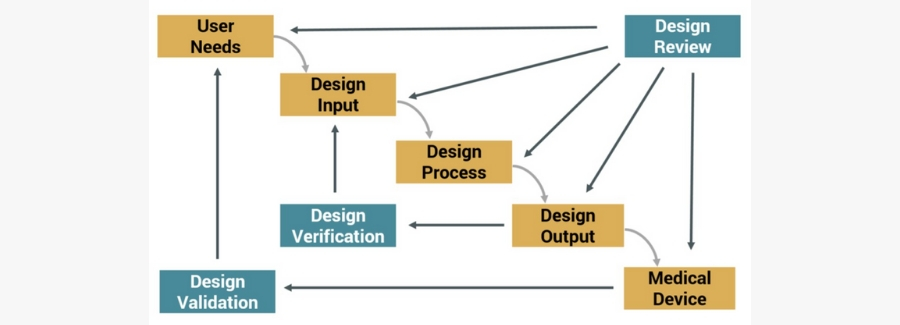
Editor’s Note: This post about getting the most out of your lab partnerships ahead up the upcoming EU MDR was originally published here on Medical Design and Outsourcing on October 5th, 2019, and was written by Joseph Tokos, Technical Director of Chemistry at WuXi Medical Device Testing.
Medical device manufacturers have a lot to keep track of these days. Between ongoing device development and preparing for the EU MDR, it’s more important than ever to maximize every partnership.
Outsourcing device testing to laboratories or contract research organizations (CROs) offers much needed relief to manufacturers and an opportunity to enhance EU MDR preparation efficiency. Here are a few insider tips to make the most of your relationship with your testing partner:
Don’t hold back on the details
Your testing partners need to ask a lot of questions about your device to be able to design a concrete test plan and accurate quote. It may seem like they’re asking for too much information before you even sign a contract, but these fine details can make or break a test plan. Failure to thoroughly detail device materials, for example, can lead to flaws in the extraction plan, such as the wrong solvent, equipment, temperature or extraction time. Complete and accurate dimensions are important because the extraction ratio will depend on wall thickness. Surface area information is also important because testing partners need to know whether the device will need to be cut into pieces for testing and whether there would be implications of doing so.
Providing all of the information upfront will save time and help prevent delays, denied submissions due to inadequate results and increased costs when studies aren’t designed properly.
Before requesting a quote, gather detailed information on the following:
- Purpose, category and patient contact time
- Size, thickness and surface area
- Materials, colorants, pigments, adhesives, additives, polymers and manufacturing aids
- Procedures used to manufacture and sterilize the device
- Parts and composition
- Existing data from previous testing
Read up on regulations
Understanding how new regulations and standards apply to your devices will empower you to educate your internal team and instill a sense of urgency to collaborate with your testing partner. Making an effort to keep abreast of the latest on EU MDR will allow you to have more productive and timely conversations about your test plans.
If you don’t have the bandwidth to dig into testing requirements — after all, that’s part of why manufacturers work with outside resources — you’ll at least want to check in with each person or department involved in device development. Design engineers and material suppliers, for example, will help you understand the intricacies of the device’s design, materials and parts.
It’s also important to note that patient contact time and in vivo testing methods will be under heightened scrutiny. Devices with shorter patient contact time are now going to be held to the same standards as longer patient contact time. This means the current data packages on even Class I devices may be incomplete moving forward. If the duration of patient contact is shorter than 30 days, be sure to ask your testing partner if you have remaining gaps in your submission that will require new testing. Also, take this opportunity to evaluate all of your options with existing in vivo device data or product family grouping data to minimize in vivo testing.
Learn from others’ mistakes
Sometimes, even the best intentions can’t guarantee a smooth testing experience. Beyond looking into what the regulations require and providing thorough product information, there are a few pitfalls that can lead to incorrect testing parameters, delays and added costs.
One common mistake is providing outdated information from a previous design. Start each device information request from scratch instead of copying and pasting from a previous form; then, investigate any gaps. This forces you to consider all the details carefully, which reduces the risk of an improperly designed test plan. Getting it right the first time reduces the risk of needing additional device samples (test article) and repeating tests.
It’s critical to provide truly representative test article of the devices produced by manufacturing. Using a prototype, for example, could yield inaccurate results and put your submission’s approval at risk because prototypes may be made of different materials or employ a different manufacturing process.
Another easily avoidable mistake is failing to describe any device changes in detail. Your testing partner can better address risks of your updated, next-gen device if they have a good understanding of what prompted the updates. Whether you addressed patient safety concerns or made updates to improve user experience, communicating the reasoning behind any device changes allows a lab to tailor the study.
Finally, don’t misunderstand the role of chemical characterization. While biocompatibility testing is important, chemical characterization is what identifies risks you didn’t even know exist.
There’s a lot to consider from a regulatory standpoint right now, and you need to maximize on every partnership to keep pace. Device manufacturers and labs alike are facing capacity constraints. If you provide a forecast to your partner and inform them of what samples they can expect and when, they may be able to reserve testing space and adjust capacity to better meet your needs. For more tips on how to prepare for MDR, read “Countdown to EU MDR: Do you know your options? ,” “Pre-clinical medical device testing under ISO 10993-1 and the MDR” and “What you need to know about reusable devices and Europe’s MDR.”