Medical technology companies, especially those developing medical devices with increasing complexity and product variations, are faced with a wide range of methods for managing risk. The toughest part is often deciding which specific methods and data points need to be captured to prove that all angles of risk analysis for product development took place. Against this backdrop of constant innovation, medical device manufacturers face unique challenges to comply with significant regulatory changes in Europe and the United States — some that could support new product development, while others could hinder it.
Changes in compliance standards, such as the new Medical Device Regulation (MDR) in Europe, include sweeping changes regarding how companies and distributors demonstrate the safety and efficiency of products. Companies are required to show clinical evidence for all products as well as provide systems and documents to remain compliant during the device lifecycle. One-way companies are reacting is by adopting modern requirement management solutions for improved agility and flexibility. With the ability to trace all aspects managed in one place, unified cloud requirement management applications can help maintain compliance and facilitate engaged collaboration across geographies. Evolving a regulatory environment means it is no longer feasible to have unrelated content without convenient access.
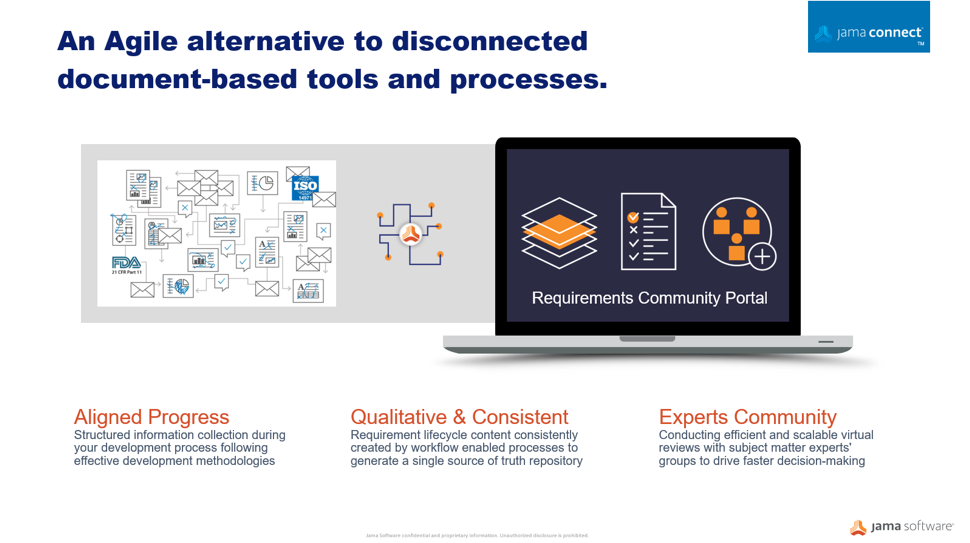
Whether regulatory authorities are tightening rules around medical devices or responding to market innovation, manufacturers must be prepared to demonstrate their products are safe and effective across the total lifecycle using a more comprehensive array of data sources. Critical factors for medical device makers must transform the way they innovate, design for manufacturability, and differentiate their products in the market. A centralized Agile alternative to disconnected, document-based processes enables pathways to faster product development, improved quality, and compliance to safety regulations.
How can you modernize and where should you start to meet these compliance challenges while streamlining your development processes and creating efficiency? Three improvement strategies can be used to insert risk control as a crucial part of the medical device development framework to comply with ISO standards and EU Medical Device Regulation (MDR).
RELATED: Getting the Most Out of Your Lab Partnership Ahead of the EU’s MDR
Three Strategies to Streamline Workflow and Strengthen Collaboration when Developing Medical Devices
With the magnitude and complexity of data expanding, medical device firms are working to assemble and analyze data faster. Much of this data is often spread out across information silos. As a result, medical device companies are increasingly looking to modern clinical requirements and safety management systems, including central data capture, that collect and clean this clinical data, all in one manageable platform.
According to a study by the Emergo Group, nearly 60 percent of medical device builders say clinical data management rules are the most challenging component of Europe’s new MDR. An equivalent percentage of companies say they don’t have a strategy for compliance. Having an approach that incorporates a complete view of data can help with the preparation and ongoing compliance. To have a plan of action, three strategies that can help you to focus on a trustful quality and efficient control of developing advanced medical products.
Establish a Single Source of Truth Framework for Reliable Insights
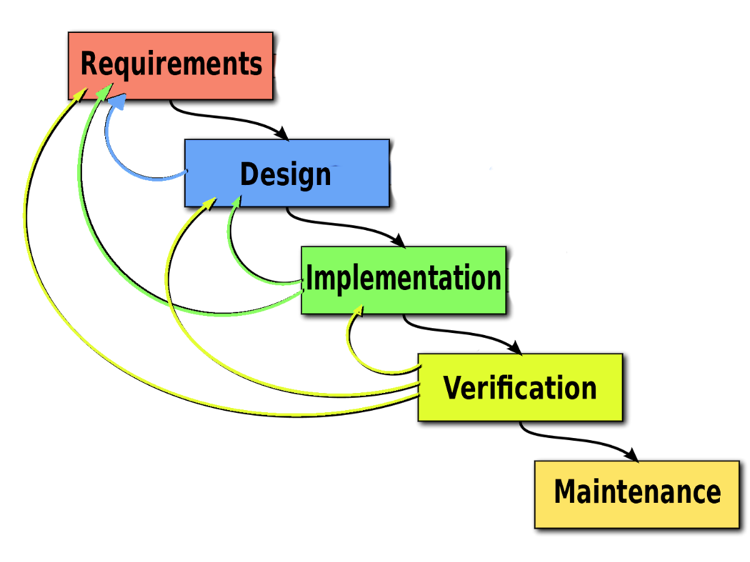
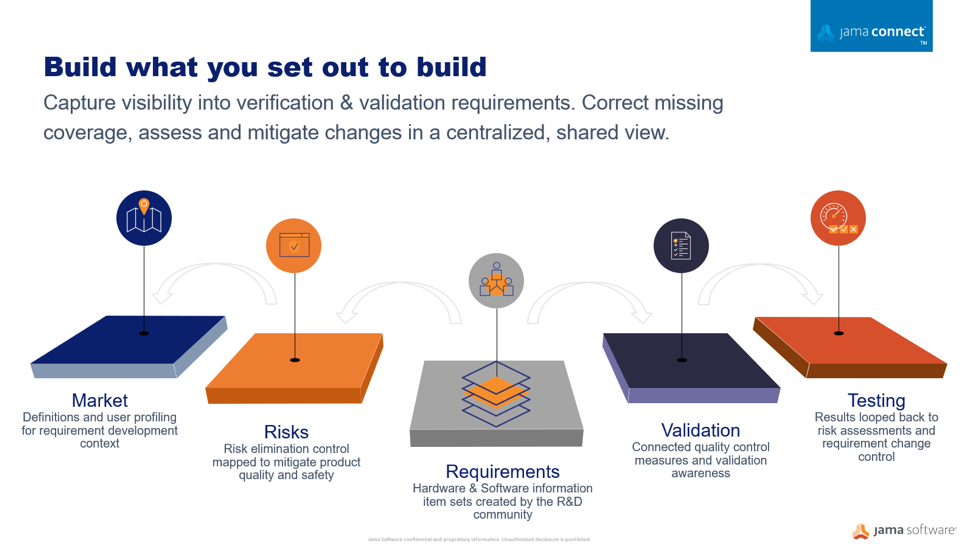
Through medical device development, many disparate documents are gathered into a collected set of information and submitted to regulators. This auditing process can be a time consuming and very manual exercise often done too late in the development cycle. Optimizing workflows to centralize information sets in an item-based approach where processes and documentation move from the document-centric model to a relational database approach can protect your quality of building new products and services. A workflow-driven, traceable methodology, and managing all information sets in real-time brings better handling to safety-controlled development cycles, and maintains a continuous state of acceptance readiness.
Streamline Risk Control with Requirement Definitions
Using a centralized information gathering approach, you can author the risk process directly connected to your development work. Your risk acceptability criteria can be defined and documented as part of the risk assessment related to the required development specifications. Moving to connected risk-requirements workflows, provides a collaborative place where the risk management plans can be viewed holistically by the entire development team in the context of the medical device development environment where requirements, specifications, validations, and risks are analyzed.
Enable Your Expert Community
Building complex products is never a one-person show. Medical device manufacturers when implementing tools, resources, and techniques to develop safe and successful products faster. A substantial factor for success is the team’s understanding and engagement at every aspect and stage of the development lifecycle, creating a robust traceable collaboration community and the ability to initiate a flexible evaluation process. These factors can have a significant impact on achieving a timely and cost-effective control that assures product quality due to a risk embedded impact analysis.
Learn more about how Jama Connect™ helps medical device developers streamline and speed up the development process while reducing risk.
Why Risk Management, Why Now?
Medical device developers can optimize the development cycle by bringing risk control and development data together in an information-centric and automated fashion and instead focus on quality, innovation, and efficiency, as well as time to release, allowing medical device builders to recognize new product revenue within shorter release cycles.
Early transitions to renewed regulations, such as the new European MDR/FDA standards, are critical in giving products a competitive advantage. Companies can turn this around into an opportunity by adhering to the following recommended steps:
- Implement a strategic, information centric approach early on to ensure market compliance and build a trustful reliable data source
- Handle your risk control elements directly connected to your traced development activities
- Make use of Jama’s consulting expertise and the available medical assets such as ISO 14971, FMEA, FTA, and 21 CFR Part 820 to rollout your renewed strategy based on industry best practices.