In this post, we’ll explore how Jama Connect for Medical Device Development is designed to help you get ramped up quickly with a platform, training, and documentation aligned to industry regulations ISO 13485:2016, 21 CFR 820.30, and ISO 14971:2019, while applying a proven systems engineering approach to product development.
With this solution, medical device teams can manage design controls for device requirements and related risks, simplifying regulatory submissions and audit preparations while accelerating time to market.
Manage Design Controls for Device Requirements and Related Risks in a Single Platform
Easily Demonstrate Traceability
Traceability ensures that design inputs have been met and verified, providing necessary evidence from the design control process. Jama Connect allows you to easily produce traceability documentation required by regulators.
Manage Risk Analysis
Manage risk analysis, aligned with ISO 14971:2019. Jama Connect helps teams identify and mitigate risks earlier in medical device development, saving teams from frustrating late-stage design changes and supporting the path to regulatory compliance.
Maintain Audit Trails and Export Data
Real-time reporting and baselining allows you to track all changes to information within the system, including timestamps and associated users. Data is easily exported from Jama Connect if your current process dictates storage of documentation in a quality management system (QMS).
Reuse and Baseline Management
Compare versions of a requirement, generate branches to develop a variant, and create catalogs of reusable requirements to improve medical device development.
Compliant Reviews and Approvals
Increase early stakeholder visibility and participation in the review process with electronic signatures that are compliant with FDA 21 CFR Part 11.
Design Verification and Validation
Seamlessly manage traceability to verifications and validations, providing evidence to comply with government regulations and standards, like 21 CFR Part 820.30.
Flexible, Scalable Options with Jama Connect for Medical Device Development
Jama Connect for Medical Device Development Solution’s license model is fully scalable, ensuring rapid deployment and easy adoption of the solution across your product development team. It also allows flexibility and can be customized to your team’s unique needs. Download this datasheet to learn more about key product benefits and features, unique licensing types, and templates templates provided as part of the solution.
To see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
https://www.jamasoftware.com/media/2020/07/2020-08-06_SolutionOverviewforMedDevice_1024x512-1.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2020-08-06 03:00:432023-01-12 16:50:25Accelerating Medical Device Development with Jama Connect
Editor’s Note: This post about getting the most out of your lab partnerships ahead up the upcoming EU MDR was originally published here on Medical Design and Outsourcing on October 5th, 2019, and was written by Joseph Tokos, Technical Director of Chemistry at WuXi Medical Device Testing.
Medical device manufacturers have a lot to keep track of these days. Between ongoing device development and preparing for the EU MDR, it’s more important than ever to maximize every partnership.
Outsourcing device testing to laboratories or contract research organizations (CROs) offers much needed relief to manufacturers and an opportunity to enhance EU MDR preparation efficiency. Here are a few insider tips to make the most of your relationship with your testing partner:
Don’t hold back on the details
Your testing partners need to ask a lot of questions about your device to be able to design a concrete test plan and accurate quote. It may seem like they’re asking for too much information before you even sign a contract, but these fine details can make or break a test plan. Failure to thoroughly detail device materials, for example, can lead to flaws in the extraction plan, such as the wrong solvent, equipment, temperature or extraction time. Complete and accurate dimensions are important because the extraction ratio will depend on wall thickness. Surface area information is also important because testing partners need to know whether the device will need to be cut into pieces for testing and whether there would be implications of doing so.
Providing all of the information upfront will save time and help prevent delays, denied submissions due to inadequate results and increased costs when studies aren’t designed properly.
Before requesting a quote, gather detailed information on the following:
Purpose, category and patient contact time
Size, thickness and surface area
Materials, colorants, pigments, adhesives, additives, polymers and manufacturing aids
Procedures used to manufacture and sterilize the device
Parts and composition
Existing data from previous testing
Read up on regulations
Understanding how new regulations and standards apply to your devices will empower you to educate your internal team and instill a sense of urgency to collaborate with your testing partner. Making an effort to keep abreast of the latest on EU MDR will allow you to have more productive and timely conversations about your test plans.
If you don’t have the bandwidth to dig into testing requirements — after all, that’s part of why manufacturers work with outside resources — you’ll at least want to check in with each person or department involved in device development. Design engineers and material suppliers, for example, will help you understand the intricacies of the device’s design, materials and parts.
It’s also important to note that patient contact time and in vivo testing methods will be under heightened scrutiny. Devices with shorter patient contact time are now going to be held to the same standards as longer patient contact time. This means the current data packages on even Class I devices may be incomplete moving forward. If the duration of patient contact is shorter than 30 days, be sure to ask your testing partner if you have remaining gaps in your submission that will require new testing. Also, take this opportunity to evaluate all of your options with existing in vivo device data or product family grouping data to minimize in vivo testing.
Learn from others’ mistakes
Sometimes, even the best intentions can’t guarantee a smooth testing experience. Beyond looking into what the regulations require and providing thorough product information, there are a few pitfalls that can lead to incorrect testing parameters, delays and added costs.
One common mistake is providing outdated information from a previous design. Start each device information request from scratch instead of copying and pasting from a previous form; then, investigate any gaps. This forces you to consider all the details carefully, which reduces the risk of an improperly designed test plan. Getting it right the first time reduces the risk of needing additional device samples (test article) and repeating tests.
It’s critical to provide truly representative test article of the devices produced by manufacturing. Using a prototype, for example, could yield inaccurate results and put your submission’s approval at risk because prototypes may be made of different materials or employ a different manufacturing process.
Another easily avoidable mistake is failing to describe any device changes in detail. Your testing partner can better address risks of your updated, next-gen device if they have a good understanding of what prompted the updates. Whether you addressed patient safety concerns or made updates to improve user experience, communicating the reasoning behind any device changes allows a lab to tailor the study.
Finally, don’t misunderstand the role of chemical characterization. While biocompatibility testing is important, chemical characterization is what identifies risks you didn’t even know exist.
https://www.jamasoftware.com/media/2020/07/2020-08-18_GettingtheMostofLabPartnershipEUMDR_1024x512.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2020-07-30 03:00:222023-01-12 16:50:26Getting the Most Out of Your Lab Partnership Ahead of the EU’s MDR
Medical device companies are on the cutting-edge of advancing health care. Along with facing relentless pressures to innovate and release quality products, they also must comply with regulations and standards to remain competitive.
On Tuesday, August 4th, we’ll be hosting a webinar specifically designed for product and engineering teams building medical devices. We’ll be covering how to move beyond the frustrations of disconnected, document-based requirement systems, streamlining your design and development and risk management processes while maintaining compliance with applicable regulations and standards.
Date: Tuesday, August 4th, 2020 Time: 8:00 – 9:00am PST
In this webinar, we’ll examine how Jama Connect for Medical Device Development helps free your teams from document-based requirements management, streamline your processes, and spend more time on innovation. Register now to learn more about:
Bringing systems thinking into design control and risk management activities using Jama Connect for Medical Device Development
Aligning how you work with the development of the artifacts needed for compliance, the Design History File (DHF) and the Risk Management file (RMF)
Defining products and risk controls through the lens of tracing
Using Jama Connect to align your design control and risk management processes with relevant parts of governing regulations and standards ISO 13485:2016, 21 CFR 820.30, and ISO 14971:2018
Presented by:
Zeb Geary – Principal Consultant, Jama Software
Zeb brings more than 10 years of experience in software consulting and business analysis to Jama’s enterprise customers and helps them deploy and optimize their use of the platform. Over the past 3.5 years, he’s worked closely with many regulated customers, especially within the Medical Device industry, to provide best practices and deployment assistance to better alignJama Connect to regulated activities pertaining to design control and verification.
https://www.jamasoftware.com/media/2020/07/2020-08_Move-from-Document-Based-Design-Control-and-Risk-Management_Social-Image-1-1.png5121024Jama Software/media/jama-logo-primary.svgJama Software2020-07-28 03:00:562023-01-12 16:50:26Medical Device Webinar: Move from Document-Based Design Control and Risk Management
As medical device manufacturers develop complex products, they require a product development approach capable of managing that complexity.
At the same time, manufacturers must continue to ensure compliance and alignment with regulations and standards. These define requirements that ensure safety and quality and reduce risk—but ultimately do not prescribe specific tools or techniques.
This is especially apparent in design control activities. Regulatory requirements define the “what” for compliance but leave the “how” to the manufacturer, as long the procedures describing that “how” remain documented and prove sufficient as part of the quality system.
This lack of a prescribed approach to managing design controls can lead to uncertainty, but Jama Connect for Medical Device Development envisions that gap as an opportunity. Here, manufacturers have the space to deploy systems engineering techniques within design control activities, supported with a robust requirements, risk, and test solution.
Note: Now that our medical device blog series is concluded, you can go back and read the series intro, Part I, and Part II.
Bring systems engineering into the design control process to manage medical device complexity.
Systems Engineering solves the problem medical device manufacturers face. It takes a complex problem and divides it into manageable units so developers can see the solution both holistically and in its interrelated parts.
Aligning to 21 CFR 820.30 and ISO 13485:2016 can naturally guide manufacturers towards this approach, requiring trace across user needs, design inputs, design outputs and verifications. The guidance does not account for the abstraction required within these areas, especially design inputs, allowing the manufacturer to determine and implement appropriate techniques and tools.
The Jama Connect for Medical Device Development solution includes:
A Procedure and Configuration Guide
An out-of-the box configuration of Jama Connect
Together, these components bring Systems Engineering principles and design control requirements into a single recommended approach.
Here’s how.
1. Apply systems thinking.
The FDA guidance (FDA, 1997) indicates that product concepts are to be “elaborated, expanded, and transformed into a complete set of design input requirements.” Jama for Medical Device Development’s procedure guide applies systems engineering principles during this design input process.
User needs are fulfilled by functions of the system.
The system allocates them to specific engineering teams or product components for further, discipline-specific definition.
In some instances, like where software is the system, abstraction of design inputs from system-level to subsystems may not cross disciplines. However, the need for hierarchical product definition remains and is reinforced by software specific standards such as IEC 62304 and IEC 82304.
2. Capture and organize design inputs.
In Jama Connect, these levels of abstractions are managed as Item Types, discrete objects within Jama Connect that allow the solution to enforce rules for how information should trace to one another.
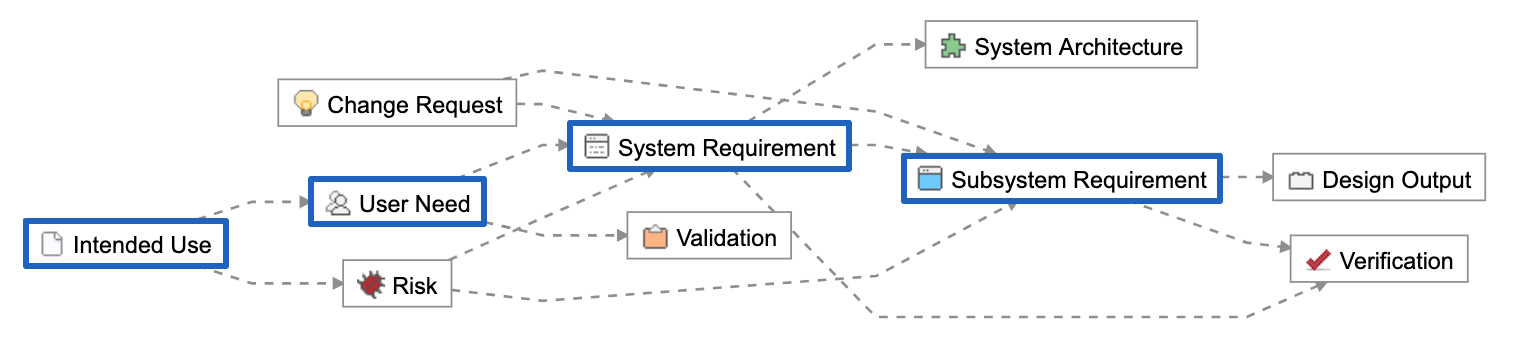
Concept-level information is captured as Intended Use and User Needs, engineering design responses as System and Subsystem Requirements. The total resulting set of requirements are referred to as the Design Inputs.
3. Establish the trace.
Below is a Relationship Rule provided as part of the out-of-the-box configuration of Jama Connect for Medical Device Development. In blue are the concept-level and design inputs:
These Item Types are unique data elements within Jama Connect. They allow us to create logical groupings we can use to manage the hierarchical levels of abstraction and to further organize across disciplines.
Standardizing engineering principles for requirements management using discrete Item Types is a valuable shift. It shows customers shifting focus from pure document generation to support for how they actually work, which can range from top-down, to middle-out, to bottom-up product definition.
The end result: Actionable information and accepted design inputs.
This shift to a focus on discrete items instead of whole documents encourages teams to act on information as it becomes ready. By capturing status within each individual item (e.g., a specific system requirement), items are independently driven from “Draft “to “Accepted.” “Accepted” indicates a requirement is ready for:
Documentation in the Quality Management System (QMS).
Further decomposition or development.
Defining Verification.
This single state can initiate several activities for other teams and does not require full document completion, which tends to restrict visibility and reduce momentum.
Although this approach encourages a shift from a document-focused product definition, in reality a document needs to be generated for the Design History File (DHF). To support these potentially conflicting needs, the out-of-the-box configuration:
Drives organization of information based on a systems engineering approach.
Uses Jama Connect’s filtering capabilities to pull together information across the project for document generation.
Using filters for document generation allows you to do more than collect different types of requirements for a single document. You can use data within the items, specifically in the Status of the items, to generate a document of only accepted design inputs. You can also take a baseline as part of the document generation process. The result is a snapshot of accepted design inputs that you can use for comparison reporting to show how design inputs may have changed over time. You can also indicate in each item’s version history which version of a requirement was included in the generated documents.
Use this Design Inputs approach with the Jama Connect for Medical Device solution to make it easier to generate documentation you need while supporting how you work with systems engineering approaches.
In the next post in this series, I’ll show you how to connect design inputs with design outputs and verifications.
Watch a demo to see key Jama Connect Medical Device Development Solution features in action and understand how teams use it to support their development process.
https://www.jamasoftware.com/media/2020/06/2020-06-11_v2_PartThree_DesignInputsForJamaConnectForMedDevices_1024x512-copy-1.jpg5121024Zeb Geary/media/jama-logo-primary.svgZeb Geary2020-06-11 15:17:542023-01-12 16:50:34Part III: Design Inputs in Jama Connect for Medical Device Development
In our experience working with more than 200 medical device developers, we’ve realized how important it is to create best practices for risk management under ISO 14971, the FDA’s mandatory standard for risk assessment throughout the product development lifecycle.
In this post, we’ll outline the main clauses of ISO 14971 and explain how Jama Connect can help medical device developers build better, safer products that satisfy ISO 14971.
What is ISO 14971?
ISO 14971 is an international standard that sees risk management as a product lifecycle process encompassing the development, production, and post-production stages. Jama Connect offers a straightforward approach to managing risk according to ISO 14971 in one platform. The standard was updated in 2019, providing more guidance on risk management and adding more detailed requirements.
Managing Risks & Requirements for ISO 14971
Risk management is an inextricable part of the medical device development process. For medical device developers, risks are a core principle of product development and should be tied together in one powerful platform.
Many medical device companies continue to depend on Excel to capture risk data, but Excel simply can’t provide the end-to-end traceability necessary for satisfying ISO 14971. That’s where Jama Connect comes in: It allows teams to easily connect risks, requirements, and testing in one system where requirements and test results stay live in real-time.
Jama Connect and ISO 14971
Jama Connect guides compliance with Clauses 4 through 7 of ISO 14971, which covers how risk should be managed throughout the product development process.
Clause 4 of ISO 14971 concerns how risk is organized and administered for your product line. It requires the formation of a Risk Management Plan throughout the development lifecycle.
The Risk Management Plan is the record of a planned process for risk management: who does what and when, how risks are scored, etc. It’s a component of the Risk Management File, which contains all the outputs for risk.
Clause 5: Risk Analysis
Clause 5 of ISO 14971 requires that medical device developers identify potential hazards and hazardous situations. Each hazardous situation and its potential consequences must be evaluated. Jama Connect helps teams satisfy Clause 5 by defining device-specific hazards and capturing risk probability and severity.
Jama Connect offers risk management item templates to capture important information about the risk analysis process, including a description of the device, intended use, and the scope of the analysis.
Teams can identify and evaluate potential hazards, sequences of events, hazardous situations, and harms in a single item type.
Clause 6: Risk Evaluation
Clause 6 requires the evaluation of risk for each hazardous situation and the definition of acceptability criteria for determining when risk reduction is required. To satisfy Clause 6, teams take the inputs from Clause 5 and determine the risk level for each hazardous situation.
In Jama Connect, risk acceptability criteria can be customized for a particular product line or medical device classification in the risk management item.
Clause 7 requires risk control measures to be developed, implemented, and verified across the product development lifecycle. Risk control measures could include product design, preventative measures in the product, and labeling. Residual risk must be evaluated against acceptability criteria, and risk control measures must be reviewed in case additional risks have been introduced inadvertently.
The risk evaluation item lets users identify risk control options for a specific hazardous situation, such as inherent safety by design, protective measures in the medical device or manufacturing process, and safety information.
Risk control measures, implementation verification, and verification of risk control effectiveness can also be accounted for in the risk evaluation item. Links to system requirements and verifications in Jama Connect can easily be created from the risk item to demonstrate traceability from hazardous situations to risk controls.
Clause 8: Residual Risk
Clause 8 requires an evaluation of the medical device’s overall residual risk. If the overall residual risk is unacceptable, it must be demonstrated that the medical benefit outweighs the residual risk.
When defining risk control measures, teams can capture those measures in Jama Connect and link them directly to risks before updating the rankings to determine the residual risk level.
With traceability through all phases of risk, users can quickly identify potential pitfalls in the product development process and address them before they become bigger barriers to success.
The Bottom Line
ISO 14971 requires a cohesive, well-documented narrative of your product’s lifecycle to assure the FDA that the device is safe, effective, and compliant. Any decisions or actions that aren’t documented could keep your product from reaching the market or result in a recall.
Finding and fixing errors early in the product lifecycle saves money, speeds time to market, and improves product quality. Jama Connect allows medical device developers to review risks and risk controls holistically so that teams can operate with confidence.
From a compliance perspective, the Jama Connect for Medical Device Development illuminates the risk management and product development process, while simultaneously generating the required documentation to support that narrative.
https://www.jamasoftware.com/media/2019/01/2020-06-08_ISO-14971.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2020-06-08 14:00:402023-01-12 16:50:35ISO 14971: Managing Risk for Medical Device Developers in Jama Connect®
Building and then bringing a medical device to market as quickly as possible—all while preserving acceptable levels of quality and regulatory compliance—requires adept medical device risk management. By minimizing potential risks such as mislabeling and software-related issues, medical device manufacturers make each product safer for the patients who will use them. All of these risks and others are present through the product development lifecycle, where they must be addressed through specific risk management activities
The ISO 14971 standard, as encapsulated in ISO 14971:2007 and then revised in ISO 14971:2019, is the modern framework for such efforts. An FDA Recognized Consensus Standard, it has a nine-part structure defining the criteria for medical device risk management during production and post-production. ISO 14971 is also required by higher-level regulation under ISO 13485. All medical device companies follow ISO 14971, but their individual approaches to the risk management standard will vary based on not only product type but the actual tools used for handling risk analysis and control measures as well.
Risks as requirements: What’s the best approach to medical device risk management?
Effective medical device risk management is integral to patient health and safety. A study published by The British Medical Journal found that one in 20 patients experiences preventable harm when receiving medical care. Moreover, many medical devices in active use are many years old, meaning that even flaws implemented long ago can continue to pose risks to patients. That means risk must be curbed at every stage of the product lifecycle.
Another way to look at it: Risks are central requirements when it comes to the medical product development process, and safely managing them in accordance with ISO 14971 requires a comprehensive modern solution capable of delivering the necessary coverage, speed and preparation. Risk management is requirements management in the medical device industry. Accordingly, it’s crucial to have the capacity to, for instance, connect eventual verification tests back to requirements, so that teams can be confident of adequate risk mitigation.
However, many existing workflows and tools cannot consistently ensure acceptable compliance, leading to the possibility of recalls, or inadequate workarounds such as alterations to the label or instructions-for-use. Spreadsheets exemplify the limitations of older approaches to requirements management and risk management during the medical device lifecycle.
The problem with document-based processes
Medical device manufacturers may rely on Microsoft Excel to capture risk data and fuel their risk management planning and reporting activities. The potential problems with this approach include:
Limited scalability to teams working at multiple locations.
Siloed data sources that take time to comb through and reconcile.
Human factors such as miskeyed entries or inadvertent deletions.
Difficulty proving compliance, due to lack of end-to-end traceability.
Taken together, these issues make it onerous to maintain and execute on a medical device risk management plan that fulfills all provisions of ISO 14971. This standard requires a combination of risk analysis, evaluation and control – all processes that a risk management plan helps simplify by documenting all of the potential risks across the product lifecycle.
How to more reliably put a risk management plan into action
The risk management planning process should produce a plan that contains product-specific data and follows all standard operating procedures in the domain. The plan should also be a living document that can be continually updated as requirements and risks evolve, as it will serve as the blueprint for ongoing risk management activities such as reporting on hazards and risk control measures and also linking back to requirements. Ensuring acceptable levels of detail and accuracy in the risk management plan is much easier with an all-in-one solution than with a collection of discrete documents.
Let’s say a hypothetical medical device company was developing an MRI machine. If it were centering its risk management processes within a massive Excel sheet, lots of valuable time would be lost to stakeholders on the development team having to constantly review the requirements in the shared asset.
Plus, this highly manual, error-prone process can itself create further complications for overall medical device risk management, such as a risk going initially overlooked due to outdated data in a spreadsheet cell. Going back later to write a report about the risks is not a great alternative to building in risk management throughout the development process—but the right tools are needed for the latter strategy.
By switching to a more modern solution, this development team could instead:
Take advantage of risk plan templates to ramp up more quickly.
See live risk mitigation data, not outdated entries.
Avoid the various administrative risks of Excel, like splitting/merging cells.
Easily adjust probabilities and severities of the defined risks.
Enable real-time collaboration between teams.
Visualize and trace risks across the whole product development lifecycle.
Prove ISO 14971 and other regulatory compliance more easily.
At the end of the development process, the team making this hypothetical MRI machine would be able to see clearly how its verification tests traced back to the risks and requirements it initially set. More specifically, they would know if the product could move the right amount of air, survive the expected transport and storage conditions and comply with all applicable rules and regulations.
Demonstrating ISO 14971 compliance with Jama Connect
Jama Connect offers a modern alternative to document-oriented processes for medical device risk management. Jama Connect is built to help streamline compliance with ISO 14971.
For example, Clause 7 of ISO 14971 requires attention to residual risks, or those risks that exist even after all risk control measures have been implemented. In Jama Connect, those measures can be efficiently defined and linked to corresponding risks for maximum traceability. That way, teams can spot potentially unacceptable risks early on in development and mitigate them before the associated costs and logistics become impractical.
There are many other features within Jama Connect for complying with all clauses of ISO 14971 and modernizing your general approach to medical device risk management to keep up with changes such as the FDA’s Safety and Performance Based Pathway. To learn more, connect with an expert today.
To learn more on the topic of risk management, we’ve compiled some helpful resources for you.
https://www.jamasoftware.com/media/2020/05/2020-05-14_WhatisMedDeviceRiskManagement_1024x512.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2020-05-14 03:00:302023-01-12 16:50:40What is Medical Device Risk Management?
Risk management plays a pivotal role in medical device development, helping ensure patient safety and product quality. Risk management is often misunderstood. It’s a process that could be cumbersome for organizations who don’t know where to get started, or worse, start too late in their development process.
After our recent half-day risk management seminar in the Netherlands, we sat down with award-winning medical device risk management educator, author, and consultant Bijan Elahi to hear his thoughts on risk management in medical device development.
Read the full interview below for Elahi’s take on the importance of risk management, best practices, and how use it to gain confidence in your compliance.
Jama Software: Thank you for joining us Bijan. For our readers who may not be familiar with your work, can you tell us a little bit about your background and experience with risk management?
Bijan Elahi: I’ve been doing systems engineering and risk management for the last 30 years, beginning my work in the aerospace industry at various companies, with my last job at NASA. In the early 1990s I was approached by the medical device industry for help with risk management. This was a time when there were no standards for medical device risk management, and medical device companies had no consistent way of doing risk management. I began helping one medical device company, then another, and soon I changed industries altogether, from aerospace to medical technologies.
The first version of ISO 14971 was published in the year 2000, finally providing risk management guidance to the global medical device community. I’ve been helping medtech companies understand that standard and apply it to product development ever since. In the last five or six years, I’ve also started teaching and consulting in risk management more broadly.
Jama Software: Looking back at your work in risk management, can you summarize for us what it is and why it’s so important to medical device development?
Bijan Elahi: Risk management, in a nutshell, is the process of identifying hazards, estimating the risks, controlling the risks, and then monitoring and reporting how you did. But once you identify the hazards, you also need to know why they happened, what are the causes of those hazards, and then what you can do to minimize the risk of harm from those hazards. Risk management is both an art and a science. It is not like mathematics that is perfectly clear. There are a lot of grey areas in risk management that require sound judgement and both a creative and a scientific approach.
Risk management is important because you can’t commercialize a medical product without it. It’s legally required to perform risk management before you can get approval to sell your product globally. Second, it helps you make safer products. We have a moral obligation to our patients to make the safest possible products for them.
It’s also important for business and financial reasons. Poor risk management can be expensive and often results in recall costs and even punitive damages. If you make safe products then you’ll have a better reputation, and you can sell more. In addition, it helps save money in product development, because risk management allows for the early identification of design weaknesses and allows targeted allocation of limited engineering resources with priority to the safety critical areas.
Jama Software: What are some risk management best practices product developers should consider as they’re moving through the development cycle?
Bijan Elahi: One of the things that medical device developers should recognize is that risk management should be done as early as possible. Even as early as the concept stage. Another thing is having a robust process for risk management that is both compliant and efficient. You can get really complex with risk management. So, avoid overcomplexity and have a process that is both understandable and explainable. Understandable to your own staff and also to regulatory bodies. When you have a regulatory auditor or reviewer asking questions about your risk management process, if you can’t help them understand, your process isn’t working. Because ultimately, you need to persuade a regulatory authority to approve your product.
Read our eBook to learn more about risk management for Class II and Class III medical device development.
Jama Software: You spoke in your presentation about an old vs. new way of thinking about risk management, can you expand on what you meant for our readers?
Bijan Elahi: The old way of thinking was where product developers saw risk management as a necessary evil. They would be focused on the design of the product and making it work. And then when they finished the design, they would say, “Well, if we’re going to commercialize this product, we need to show that we did risk management, and find somebody to do a retrospective analysis and write a report that this product is adequately safe.” That’s old thinking. The new way of thinking is to consider risk management as a value-added activity and a partner with product design. Risk management and product design should work together in lockstep. This way, we get real value from risk management.
In fact, regulatory bodies today expect this new way of doing the work. They don’t want to see that you didn’t do any risk management during the development process and that at the end you just wrote a report to retrospectively cover your bases. This way of thinking is not only bad for your company, it’s also bad for the consumer and bad for the product. A lot of times if you have finished your design, and then suddenly you make a discovery of a safety hazard, it may be too late to fix it or maybe just too expensive. Some companies would just try to somehow get the product out anyway, which is bad for everybody and doesn’t really save you any money.
Jama Software: Let’s talk a little bit about the latest version of ISO 14971 that was released at the end of last year. Have you had a chance to review the latest 2019 version and are there any new changes developers should be aware of?
Bijan Elahi: Yes, I have. ISO 14971 is the international standard for medical device risk management, and it is recognized by most countries. It is a very important standard because conformance to it is the easiest way to establish the safety of your medical device and to persuade a regulatory body to approve your device. The 2019 version has some changes in it, but they are not so extensive that they would overburden the manufacturers. There are some new definitions and some changes to existing definitions.
Also, there are some new concepts that are introduced. For example, in the 2012 version, the requirement was to reduce the risks “as far as possible.” In 2019, a new concept is introduced to reduce the risks to “as low as reasonably achievable.” And then in the previous version, 2007, there was another concept called “as low as reasonably practicable.” So, there are now three concepts in the most recent version about how can you strategize about reducing risks.
Jama Software: Speaking of changing industry regulations, one topic that’s been top of mind for medical device developers in Europe is the upcoming EU Medical Device Regulation (MDR). Is there anything that developers should consider when getting ready for MDR?
Bijan Elahi: Yes. MDR really created a ground shift for the medical device manufacturer. It caused a lot of change, and it’s more of a quality regulation. Some of the biggest changes with respect to risk management are the requirement to submit an annual Periodic Safety Update Report (PSUR). The PSUR is for Class II, and above medical devices. Another requirement is Post-Market Surveillance Reports (PMSR) for Class I devices. There will also be more emphasis placed on post-market clinical follows ups. The regulators expect you to continue to follow up your product and to see how it is clinically performing.
MDR puts more emphasis on post-market surveillance plans. You’ll need a plan ahead of time for surveilling your medical device and how it performs in the field. You’ll also need to identify and declare the lifetime of the medical device and provide a rationale. Article 88 in the MDR also requires trending, which will result in more reporting required by the manufacturer. If an adverse event hasn’t happened yet, but looks like it’s going to happen, then you have to report that too.
Jama Software: Interesting. So, you need to anticipate risks before something happens and there will be more documentation required?
Bijan Elahi: Yes. Before, we had vigilance reporting. Which means something bad has already happened, and you had to report it to the governmental agencies in Europe. Now in addition to vigilance, you have to predict if something bad is going to happen and report that as well.
Jama Software: Do you foresee any changes to risk management or requirements management being impacted by MDR?
Bijan Elahi: Good requirements management is just so smart to do for better and more efficient product development. I use it in risk management as well.
Basically, I treat hazards, risk controls, and safety requirements all as elements in the requirements management system. So, they’re all connected and traced. Traceability is another thing that is really emphasized in ISO 14971. It is so easy to do traceability with a requirements management solution like Jama software and, so hard to do it manually.
If you try to do it by manually, e.g., with an Excel spreadsheet, it’s so easy to lose traceability and have errors. It’s also hard to maintain. Anybody who has done traceability analysis knows how much work it is and how hard it is to maintain it because designs are dynamic. Things change all the time. Anytime you make a change, you could break your traceability. This is one thing that I always advise my students and my clients, to use a tool like Jama Connect.
Jama Software: Are there any specific best practices or any highlights that you can address in regard to risk and requirements management together? In regard to the discipline of combining those two and how they can help product development and launching products.
Bijan Elahi: Yes, I am a strong believer that risk-management and requirements-management are better together. For example, safety requirements: How do you know which requirements are safety requirements? A safety requirement is that which is linked to a risk control. As risk controls themselves are elements in the database, if you link some of your requirements to those risk controls then you can tag those requirements as safety requirements.
Sometimes when you are audited, an auditor asks you, “What are your safety requirements?” With the explanation that I just gave, it is very easy to run a query with a solution like Jama Connect for all of the requirements that are tagged as safety and get a list of them. Very easy to do! Without a tool, it’s not so easy to answer that question.
Another example is connecting Failure Modes and Effects Analysis (FMEA) analysis to your risk management. The tool can help manage the connections of the causes and effects that lead to hazards. Again, those causal factors themselves could be elements in a database which are linked together and that creates what’s called a sequence of events, which explains how a hazard can manifest itself. By creating this chain of events, you can connect the hazard to the hazardous situation, and subsequently to the harms. You can do a quantitative computation of risk, if you follow this methodology.
Jama Software: What are some common mistakes that you’re seeing medical device developers make in their risk management process?
Bijan Elahi: The first mistake that comes to mind is actually just an approach that is suboptimal. One of the things that many device manufacturers do when it comes to assignment of severity of harm is assign just one classification to it. For example, if you are infected, they would ask, “What’s the severity of the infection?” And they would assign a severity classification like serious or critical. These are key words that I’m using, by the way.
The thing is that a harm doesn’t create just one kind of outcome, but multiple outcomes. For example, an infection could cause anything from just a little bit of a fever, to organ failure, to even death. So, it’s not best practice to assign only one severity class to a harm. It’s best to assign a spectrum of severity classes to harms.
Jama Software: As we enter a new decade and medical devices are becoming more complex and more embedded in our everyday lives, how do you think risk management might change?
Bijan Elahi: Well, I think risk management is going to become more and more important. Especially with the introduction of EU MDR. EU MDR puts more emphasis on risk management. I see a lot more ‘risk-based’ language everywhere. Decisions need to be risk-based. That applies to all kinds of decisions, including design changes and verification testing. If you’re going to make ‘risk-based’ decisions, you need to know what risk is. And where you get risk information: is from risk management. Risk management is a progressively more prominent endeavor in medical device development.
The new regulation, which emphasizes patient safety, traceability, and transparency, will impact medical device developers, manufacturers, and suppliers of all sizes across Europe, and will require fundamental changes throughout the entire device lifecycle.
In order to better understand the regulation and its impact on our medical device customers, we sat down with Satyajit Ketkar, Principal System Architect & Engineer at Velentium, to learn more about what MDR is, what medical device developers can expect, and what they should be doing to prepare prior to the May 26, 2020 implementation date.
Jama Software: What is EU MDR and what can medical device developers in Europe expect?
SatyajitKetkar:
MDR stands for Medical Device Regulation. It combines and replaces the existing directives. Once the MDR goes into effect (on May 26, 2020), the existing directives will be obsoleted and invalid. All medical device manufacturers will be required to comply to the MDR. Right now, there are three different directives: The Medical Device Directive (MDD), the Active Implantable Device Directive (AIMDD), and the In Vitro Diagnostic Devices Directive (IVDD) — The EU governing body took two of those —the MDD and the AIMDD — combined them together and brought it to the next level to make it a regulation, which means that it’s extremely explicit and binding. There’s also the IVDDR, like the MDR and will replace the IVDD. The MDR is also harmonized with a lot of the latest standards. EU MDR is supposed to provide the medical device manufacturers a much more comprehensive regulatory guidance on how to manufacture a medical device. But it goes way beyond that, it goes from phase-zero feasibility, all the way through end–of–life (EOL) of the device. There’s a lot of emphasis on clinical, post-market surveillance, how to handle complaints, auditing, review cycle – whether it’s three-year or five-year. There’s a lot more content in there. It’s a pretty large document. It’s supposed to be in the same line as the data protection regulation (GDPR).
Jama Software: Why are they implementing new regulations around medical device development?
Satyajit Ketkar:
They’re basically pushing these regulations up to a level where the ambiguity and the ‘legalese’ is taken out. In the past, a directive was set out through the EU legislative body from Brussels, and then each competent authority for each country would interpret it in their own way. Each of the notified bodies, like BSI, TÜV, or DEKRA, had their own interpretation of the directive. So, if you’re a legal manufacturer, you could go to each one of these bodies and get a different implementation of the directive. What this new EU MDR regulation is supposed to do is to clear that up. Everybody now operates in the same manner, in the same language. And because of that, the regulation is large.Whereas the MDD about 60 pages and has 23 articles and 12 annexes, the MDR is about 175 pages and consists of 123 articles and 17 annexes.
Jama Software:
What are the top things that you suggest medical device companies do to prepare for the new regulation?
Satyajit Ketkar:
Overall, one of the first things I would recommend any medical device manufacturer do is to read the articles that talk about device classification and understand where they fit in.
If you’re an existing manufacturer of a product that’s already out in the market that falls under either the MDD or AIMDD, you’ll need to understand where you fall on the new spectrum. You must understand if your medical device still falls under the same classification, or if it has changed.
Most likely, they’ve gone up in classification. If they were Class 1R or Class 1M, now they’re Class 2A. If they were Class 2A, they’re now Class 2B. If they were Class 2B, they’re now Class 3. And of course, the current or future AIMDsremain as Class 3.
So, what does that mean? That means that the rigor of their development processes and the evidence that they mustmaintain, the DHF (Design History File) records and what they mustprovide has just gone up by quite a bit. When these directives were originally written back in the ‘90s, a lot of the technologies we’re working with today didn’t exist. So, to capture a lot of that, they have up classified (changes to the rules for a device classification, see above).This is especially true for electrical hardware, firmware, software, cybersecurity, and the mobile applications.The rules and expectations for these areas have gotten more explicit and stricter.
It seems like a big part of MDR is that there’s going to be more traceability needed, unique identifiers, and more labeling for each device. Do you see that impacting the way organizations develop medical devices? And what’s the best way to capture that information, or anything else related to post-market surveillance?
Satyajit Ketkar:
So, for the UDI implant card, etc., that’s new, and wasn’t there before. So, everyone’s starting from scratch.
Some people were kind of doing UDI (Unique Identification Number) labeling in the Instructions for Use (IFU) or maybe the eIFU, but it wasn’t to this level of rigor. You must now maintain vigilant records for all of this, for the entire life of the product including part of the post-market. There’s supposed to be a whole database that they’re putting together to keep track of all the vigilant reporting and the post-market reporting, that’s called EUDAMED. There are now updates to the Med Dev guidance(s) on clinical and post-market that are going to be referred to within the regulation. So, I would recommend that manufacturers continue to follow and comply to that. Beyond that, it’s pretty much the same thing.
What’s changed significantly is the clinical follow-up. There’s a whole new guidance on clinical safety and performance that they must maintain, which is part of this UDI tracking and vigilant tracking. Also, there is a periodic clinical evaluationthat mustbe conducted. There’s aperiodic safety and performance report as well that needs to be updated thatmust be included in the vigilance and post-market surveillance. These, and the UDI, are all new with MDR. Before, you would do this once in your three or five-year review cycle, and then that was it. Now, you must basically maintain these all the time as part of your post-market surveillance.
This doesn’t introduce a development burden, but it is a lifecycle burden. You must maintain these throughout the life of the product, which could be 15, 20 years, especially if it’s an active implantable, a pacemaker, or any sort of a therapy device.
One of the other things that I would recommend on the regulatory side or the system side is to review Annex 1, which contains the safety and performance requirements called GSPR (General Safety and Performance Requirements). The MDD has a set of 13 EssentialRequirements (ERs), and the AIMDD has16. The MDR has 23 GSPRs. This might seem less overall but each one of these requirements has 10 to 30 sub-sections that you must adhere to. So, in reality, the requirements have more than doubled.
Typically, what happens is that when somebody in a regulatory group or clinical group puts together their submission package, they’ll review all these essential performance and safety requirements to make sure that they’ve got all the documentation or all the deliverables. They’ve gotten very explicit as to what you need to provide. One example, specifically Section 17.2, which talks about electronic programmable systems and software. They’ve gotten specific about cybersecurity. They’re catching up with the FDA on cybersecurity and software lifecycle. That’s just one example, but there’s a whole bunch of other ones in there. These are new. So, that is something that they need to be aware of as they do their preliminary risk assessment and overall risk managementto comply with ISO 14971.These will be additional inputs for decomposing their system requirements. They need to be aware of the new and existing requirements.
Learn how Jama Software can help medical device developers better manage risk with ISO 14971 by downloading our white paper.
Jama Software:
Do you think MDR will have an impact on the way people create and approve requirements? Do you have any thoughts on how the requirements managementprocess might change?
Satyajit Ketkar:
I’m moderately confident that when it comes down to a very specific phase(s) of the development lifecycle, where you’re decomposing requirements and creating your traceability, from design, implementation, through to verification, that it should stay the same. It’s just the details of what you’d have to maintain that are going to change. The process should stay the same. Because remember, there are two parts of what you do: You create a DHF to maintain for yourself as a manufacturer, and then there’s a subset of that that you send to a notified body. The burden of what you must maintain for your own records goes up quite a bit. The process stays the same. It’s just what you must keep track of now that has gone up. The traceability is going to go up a little bit, but the process of decomposing requirements to subsystem, technical, and doing the architecture design, etc., all the current and standard processes should stay the same.
Jama Software:
Do you see this impacting the way global companies communicate or collaborate as part of MDR?
Satyajit Ketkar:
Yes: it’s going to require quite a bit more communication. You must stay aligned a lot more closely because there’s a lot more burden. There’s a lot more meat on the bone you must provide. So, if you have a distributed team across multiple regions, you need a central repository to maintain all these documents.
Jama Software:
You mentioned the DHF and that burden of proof is going to really be on the company.Does it impact the way folks adhere to ISO 13485 or Quality Management Standards at all?
Satyajit Ketkar:
No, it should not. Since it is a brand-new regulation, one of the things they’ve done with the MDR is adopt all the latest harmonized standards. That includes the latest QMS standard, latest risk management, etc. So, if you’re up to date on those, then you should be fine. You should not need to change your QMS to adhere to the MDR as long as you are maintaining the state-of–the–art.
Jama Software:
You mentioned doing earlier risk management followingISO 14971. Are there any impactsto the risk management process?
Satyajit Ketkar:
No. It’s kind of the other way around. MDR has taken account for the updates to all these other standards, especially the updates to risk and QMS. So, if you follow the MDR and you’re up to date on your revision of these other standards, you should be fine.
Jama Software: Is there anything else that you think we missed, or you think should be top-of-mind related to MDR?
Satyajit Ketkar:
I want to be very specific about what I just said earlier in following the harmonized standards and maintaining the state-of-the-art. If you’re not up to date on the latest standards, for example, if you’re not following ISO 14971:2012, or if you’re not following QMS:2016, if you are behind and you want to jump up to the MDR, you’re going to have a pretty heavy lift because you must comply to the latest revisions as well. It’s all included in the harmonized standards list now as this list has been updated. So, if you’re already doing it (not just complying to the current harmonized standard but aware of the state-of-art), then you’re fine. If you’re not, there’s going to be some uphill climbing that may be required.
What’s typical in my experience is that people try to keep up with the standards more often than the regulations. But they’ll see a big jump in the regulation, and they don’t want to jump straight into the regulation and find out that they’re still back in 2006, they haven’t revved up to the latest one (typically the state-of-the-art), and they’re stuck. So, just want to make sure that manufacturers are aware of that.
Jama Software: How do you think a dedicated requirements management platform like Jama Software fits into all of this?
Satyajit Ketkar:
If you’re already using Jama Software, you won’t have to change the process too much. The only thing that this new regulation is going to change is that people are going to want to use Jama Software more because it’s going to become necessary. There’s just too much from a QMS perspective and a development process perspective to try to do manually.
Because of this new regulation, it’s going to slow down their development lifecycle at first, because they are now learning about this new regulation. On top of that, if you’re doing things manually, if you’re using Excel spreadsheets or Word documents, whatever it may be… You might have a very simple project, but now you must add that manual process burden on top of this new regulation. So, it would reduce that process burden greatly if you had a solution like Jama Connect that would handle a lot of this for you. If you could just enter in all your requirements into a lifecycle management tool that then allows you to create trace tables, allows you to create upward, downward decomposition, and presents that in a very clean way where you can export it to a DHF artifact. That saves time and effort.
That’s the whole point, is to save time, because you already are in a process and a regulation change.Tools like Jama Connect will accelerate that because I do foresee a major slowdown in the lifecycle of medical devices with this new regulation… especially for startups. They’re going to have a tough time picking this up. So, to have a platform like Jama Connect out there to aid in this is going to be very important.
Jama Software: What resources are available to people that you know of to help them comply with the new standards?
Satyajit Ketkar:
Most of the approved, certified, notified bodies in Europe — some of the ones that I mentioned before like BSI, TÜV SÜD, DEKRA — are a great resource. They should have a quick cheat sheet on their website. You can also go online to any of the medical device journals, and they have a pretty good high-level breakdown of EU MDR. It’s more on the regulatory side than a development-process side, but it gives you the breakdown of what articles are necessary, what annexes need to be reviewed, and what order to do it in.
https://www.jamasoftware.com/media/2020/01/EU_MDR_Blog.png5121024Satyajit Ketkar/media/jama-logo-primary.svgSatyajit Ketkar2020-02-11 04:30:432023-01-12 16:50:46An Interview with Velentium: How Medical Device Developers Can Prepare for EU MDR
Medical technology companies, especially those developing medical devices with increasing complexity and product variations, are faced with a wide range of methods for managing risk. The toughest part is often deciding which specific methods and data points need to be captured to prove that all angles of risk analysis for product development took place. Against this backdrop of constant innovation, medical device manufacturers face unique challenges to comply with significant regulatory changes in Europe and the United States — some that could support new product development, while others could hinder it.
Changes in compliance standards, such as the new Medical Device Regulation (MDR) in Europe, include sweeping changes regarding how companies and distributors demonstrate the safety and efficiency of products. Companies are required to show clinical evidence for all products as well as provide systems and documents to remain compliant during the device lifecycle. One-way companies are reacting is by adopting modern requirement management solutions for improved agility and flexibility. With the ability to trace all aspects managed in one place, unified cloud requirement management applications can help maintain compliance and facilitate engaged collaboration across geographies. Evolving a regulatory environment means it is no longer feasible to have unrelated content without convenient access.
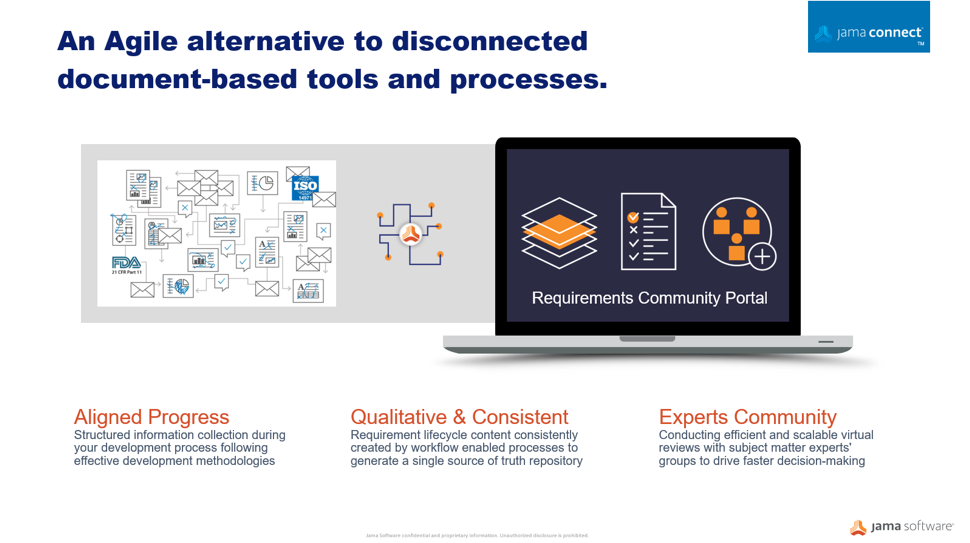
Whether regulatory authorities are tightening rules around medical devices or responding to market innovation, manufacturers must be prepared to demonstrate their products are safe and effective across the total lifecycle using a more comprehensive array of data sources. Critical factors for medical device makers must transform the way they innovate, design for manufacturability, and differentiate their products in the market. A centralized Agile alternative to disconnected, document-based processes enables pathways to faster product development, improved quality, and compliance to safety regulations.
How can you modernize and where should you start to meet these compliance challenges while streamlining your development processes and creating efficiency? Three improvement strategies can be used to insert risk control as a crucial part of the medical device development framework to comply with ISO standards and EU Medical Device Regulation (MDR).
Three Strategies to Streamline Workflow and Strengthen Collaboration when Developing Medical Devices
With the magnitude and complexity of data expanding, medical device firms are working to assemble and analyze data faster. Much of this data is often spread out across information silos. As a result, medical device companies are increasingly looking to modern clinical requirements and safety management systems, including central data capture, that collect and clean this clinical data, all in one manageable platform.
According to a study by the Emergo Group, nearly 60 percent of medical device builders say clinical data management rules are the most challenging component of Europe’s new MDR. An equivalent percentage of companies say they don’t have a strategy for compliance. Having an approach that incorporates a complete view of data can help with the preparation and ongoing compliance. To have a plan of action, three strategies that can help you to focus on a trustful quality and efficient control of developing advanced medical products.
Establish a Single Source of Truth Framework for Reliable Insights
Through medical device development, many disparate documents are gathered into a collected set of information and submitted to regulators. This auditing process can be a time consuming and very manual exercise often done too late in the development cycle. Optimizing workflows to centralize information sets in an item-based approach where processes and documentation move from the document-centric model to a relational database approach can protect your quality of building new products and services. A workflow-driven, traceable methodology, and managing all information sets in real-time brings better handling to safety-controlled development cycles, and maintains a continuous state of acceptance readiness.
Streamline Risk Control with Requirement Definitions
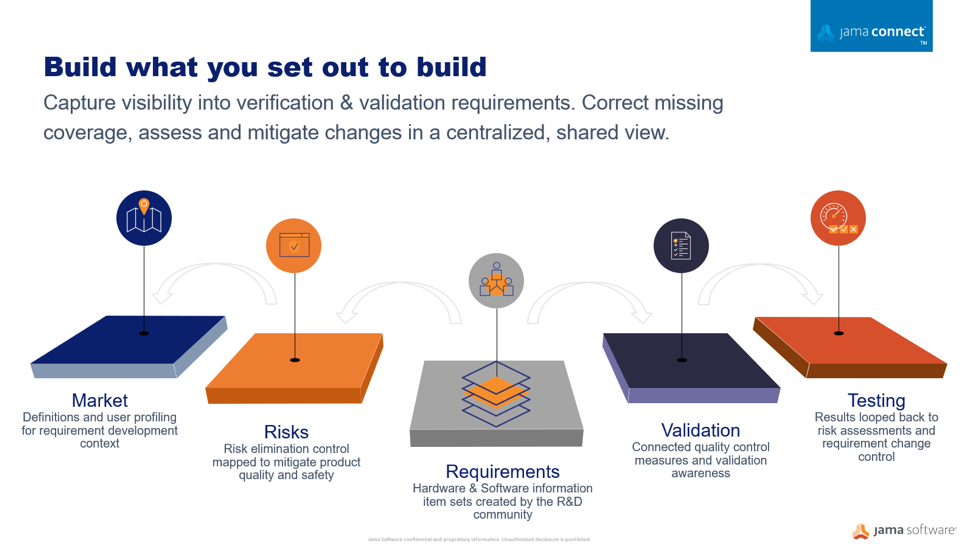
Using a centralized information gathering approach, you can author the risk process directly connected to your development work. Your risk acceptability criteria can be defined and documented as part of the risk assessment related to the required development specifications. Moving to connected risk-requirements workflows, provides a collaborative place where the risk management plans can be viewed holistically by the entire development team in the context of the medical device development environment where requirements, specifications, validations, and risks are analyzed.
Enable Your Expert Community
Building complex products is never a one-person show. Medical device manufacturers when implementing tools, resources, and techniques to develop safe and successful products faster. A substantial factor for success is the team’s understanding and engagement at every aspect and stage of the development lifecycle, creating a robust traceable collaboration community and the ability to initiate a flexible evaluation process. These factors can have a significant impact on achieving a timely and cost-effective control that assures product quality due to a risk embedded impact analysis.
Medical device developers can optimize the development cycle by bringing risk control and development data together in an information-centric and automated fashion and instead focus on quality, innovation, and efficiency, as well as time to release, allowing medical device builders to recognize new product revenue within shorter release cycles.
Early transitions to renewed regulations, such as the new European MDR/FDA standards, are critical in giving products a competitive advantage. Companies can turn this around into an opportunity by adhering to the following recommended steps:
Implement a strategic, information centric approach early on to ensure market compliance and build a trustful reliable data source
Handle your risk control elements directly connected to your traced development activities
Make use of Jama’s consulting expertise and the available medical assets such as ISO 14971, FMEA, FTA, and 21 CFR Part 820 to rollout your renewed strategy based on industry best practices.
To see more information specific to the medical device development industry, we’ve compiled a handy list of valuable resources for you!
https://www.jamasoftware.com/media/2020/01/Medical_Device_Development.png5121024Rene van Popering/media/jama-logo-primary.svgRene van Popering2020-01-28 13:16:022023-01-12 16:50:47Accelerate Medical Device Development with Modern Requirements Management
So, what is this Safety and Performance Based Pathway? Why did the FDA create it? How might it affect medical device development? Let’s examine those questions.
What is the Safety and Performance Based Pathway?
The existing Abbreviated 510(k) Program, established in 1998, was designed to lower the regulatory burden on both manufactures and the FDA and speed new medical devices to market. It permits manufactures to gain FDA approval by demonstrating their new device provides equal or better performance than an approved device that’s currently on the market (a so-called, “predicate device”).
The new Safety and Performance Based Pathway will allow manufacturers to gain approval by demonstrating that their new device meets a set of FDA-identified safety and performance criteria for its device category, rather than making a direct comparison with a predicate device.
The scope of this new pathway is limited. Issued on February 1, 2019, the guidance document states that this pathway applies only to specific device types the FDA understands very well and for which it has issued safety and performance criteria.
To that end, the FDA has begun releasing and soliciting industry feedback on guidance for specific device categories, starting with four documents issued on September 19, 2019. In each document, the FDA identifies the intended use and design characteristics of the device class along with the testing performance criteria for the class. The four device types for which the FDA has issued guidance are:
Conventional Foley catheters
Cutaneous electrodes for reporting purposes
Orthopedic, non-spinal, metallic bone screws
Spinal plating systems
The FDA plans to expand the program by rolling out many more such documents in the coming months.
Why has the FDA created the Safety and Performance Pathway?
The new pathway is part of the FDA’s effort to reduce the regulatory burden on both medical device manufacturers and the agency while speeding safer, more effective products to market with FDA approval. With the Safety and Performance Pathway, the FDA hopes to reduce manufacturer’s dependence on direct comparison with predicate devices, especially older predicates that rely on dated technologies. According to the agency, of the 3,173 devices cleared through the Abbreviated 510(k) pathway in 2017, roughly 20% were predicated on devices that were more than 10 years old.
When the new pathway was first announced in November 2018, FDA Commissioner Dr. Scott Gottlieb and Dr. Jeff Shuren, director of the FDA’s Center for Devices and Radiological Health (CDRH) said in a press release that, “Rather than looking to the past as a baseline for safety and effectiveness — and rely on predicate devices that are sometimes decades old as our point of comparison — our premarket review would be based on a contemporary baseline that looks to the future and a baseline that can be updated as technologies advance.
“Through this new path, a company would demonstrate that a novel device meets modern performance-based criteria that have been established or recognized by the FDA and reflect current technological principles,” they wrote. “These criteria would reflect the safety and performance of modern predicate devices. We’d like this efficient new pathway to eventually supplant the practice of manufacturers comparing their new device technologically to a specific, and sometimes old, predicate device.”
The press release also stated that the agency is investigating whether it can retire older devices as eligible predicates. Such a move could require Congressional approval, however.
“To be clear, we don’t believe devices that rely on old predicates are unsafe, or that older devices need to be removed from the market,” Gottlieb and Shuren wrote in their statement. “However, we believe that encouraging product developers to use more modern predicates would give patients and their doctors a choice among older and newer versions of a type of device, promote greater competition to adopt modern features that improve safety and performance, and help make sure that newer devices reflect more modern technology and standards that can improve patient care and outcomes. It would help the overall product environment continue to evolve in the direction toward more modern performance standards.”
Learn how Jama Software can help medical device developers better manage risk with ISO 14971 by downloading our white paper.
Pushback from Medical Device Industry Lawyers
Some think that with their stated goal of encouraging innovation, the FDA is overstepping its authority.
In an email to MobiHealthNews, Bradley Merrill Thompson, a lawyer who counsels on FDA regulatory issues, said phasing out old devices would be “essentially forcing companies to innovate regardless of whether the marketplace perceives a need,” thus driving unnecessary R&D and higher healthcare costs. He believes that if the FDA feels it needs to drive innovation through new regulatory changes, it would need to convince Congress to update the Medical Device Regulation Act of 1976.
“The 1976 statute is very clear, and FDA can’t simply ignore it,” Thompson wrote. “If FDA wants to change the statute, then to Congress they should go. Congress in turn could bring together all of the various stakeholders including patients, healthcare professionals and medical device manufacturers to talk about what they need and the best way to do this. FDA has no mandate to do this on its own.”
Possible Effects on Medical Device Development
Thompson’s legal concerns seem to apply only to the FDA’s investigation into retiring older predicate devices from the original Abbreviated 510(k) Program, however. From an engineering standpoint, the new Safety and Performance Based Pathway appears to offer manufacturers some important benefits.
First, the alternative pathway will allow manufactures to focus on surpassing safety and performance criteria rather than on making a comparison with a predicate device. Designers will have greater liberty to move away from predicate designs and technologies, since there is no need to make a direct comparison with a predicate device.
Second, for new devices in the FDA’s “well-understood” categories, the new pathway should speed time to market. The impact will be limited initially, but it should increase as guidance for more device types is released.
Finally, by establishing modern performance benchmarks for FDA approval that can be updated over time, the new pathway should help increase the safety and performance of new medical devices, at least those in designated device categories.
As we look to the future of medical device development, we can expect to see additional and extended guidelines from the FDA and other governing bodies on how to safely bring new products to market in a way that enables innovation. Stay tuned.
https://www.jamasoftware.com/media/2019/11/FDA_Approval_Medical_Device_Development.png5121024Jama Software/media/jama-logo-primary.svgJama Software2019-11-21 04:30:422023-01-12 16:51:16How the FDA’s New Safety and Performance Based Pathway Could Affect Medical Device Development


 Medical device companies are on the cutting-edge of advancing health care. Along with facing relentless pressures to innovate and release quality products, they also must comply with regulations and standards to remain competitive.
Medical device companies are on the cutting-edge of advancing health care. Along with facing relentless pressures to innovate and release quality products, they also must comply with regulations and standards to remain competitive.