Incorporating Design Controls can be a daunting task for medical device product development teams. For a variety of reasons, including unfamiliarity of regulatory expectations, inexperience with implementation, or viewing them as a burden preventing innovation, too many teams incorporate Design Controls reactively instead of proactively.
Here are three ways to proactively and practically incorporate design controls in your medical device development process that results in better market adoption, faster time to market, and regulatory compliance.
1. Plan for it
Design controls is not a singular activity or simple set of static deliverables that can be accomplished at the last minute successfully. Planning how your organization will incorporate design controls throughout the product development life cycle is key for a successful medical device product. At the highest level of planning is an organization’s standard operating procedure for device development that incorporates the major aspects of design controls. At the next level is the project plan for the development of a particular medical device. The complexity of the plans is scaled with the risk and complexity of the medical device being developed. Higher risk and higher complexity devices require higher sophistication of planning processes and tools than lower risk and lower complexity devices.
2. Incorporate risk management as part of your requirement setting process
Risk management is expected to be incorporated throughout the product development life cycle. One best practice is to perform the application risk analysis concurrently with determining user needs and the top level design inputs; not after development as a check-the-box activity. As risks associated for foreseeable misuse are uncovered, the team can proactively decide how the risk will be addressed, whether safety by design, protective measures, and/or information for safety and create a corresponding user need and/or design input. Now the team has a comprehensive list of requirements at the beginning of detailed design and development, not at the end.
3. Organize user needs, design inputs, and risk management information
Maintaining and organizing user needs, design inputs, sub-requirements, and individual risks is key to proactively incorporating design controls. In addition to the high number of items, there is the common scenario of many-to-one or one-to-many relationships that must be kept clear as well. For example, one design input can address multiple user needs or vice versa.
Thus, without proper organization, it is possible to miss creating a corresponding design input to a user need, linking a risk item to a corresponding design input, or missing the actual development of a design input during the development phase; all leading to unwanted, reactive last-minute development efforts. In addition, once the user needs, design inputs, and risk management are well organized, tracing the corresponding design verification and design validation efforts is much easier.
Choosing the appropriate tool can simplify these requirements management efforts. For lower complexity devices, management of design inputs and risk may be sufficiently organized using a word document or spreadsheet. Note that even a ‘simple’ device can create a list of requirements and risks of notable length. I’ve worked on a plastic sub-assembly that sat over a nasal spray pump as part of a drug-device combination device. With no mechanical moving parts or software, the list of design inputs still spanned a dozen pages, and the risk analysis FMEAs numbered hundreds of line items.
As the medical device or technology being developed increases in complexity, so does management of the requirements and risks. In contrast to a drug-delivery sub-assembly, I’ve also been involved with the development of a robotic surgical system. With a handful of sub-systems, embedded software and software user interface, the resulting requirements, sub-requirements, and sub sub-requirements were a challenge to manage via simple word docs and spreadsheets, to say the least. These days, software tools such as Jama Connect® allow for visual management and traceability of user needs, design inputs, risk management, design outputs, design verification, and design validation. With all items in an integrated platform versus disparate spreadsheets and documents that are linked manually, teams can spend less time on the requirements management, thus leading to faster, better product development with high confidence in meeting regulatory compliance.
These are just three of the ways to integrate design controls proactively into your organization’s medical device product development process. Proactive integration done practically adds value and enhances, not hinders, innovation.
https://www.jamasoftware.com/media/2021/09/2021-09-27-proactive-vs-reactive-med-device_1024x512.jpg5121024Michelle Wu/media/jama-logo-primary.svgMichelle Wu2021-09-27 03:00:482023-01-12 16:48:04Three Ways to Proactively (vs Reactively) Incorporate Design Controls in Medical Device Product Development
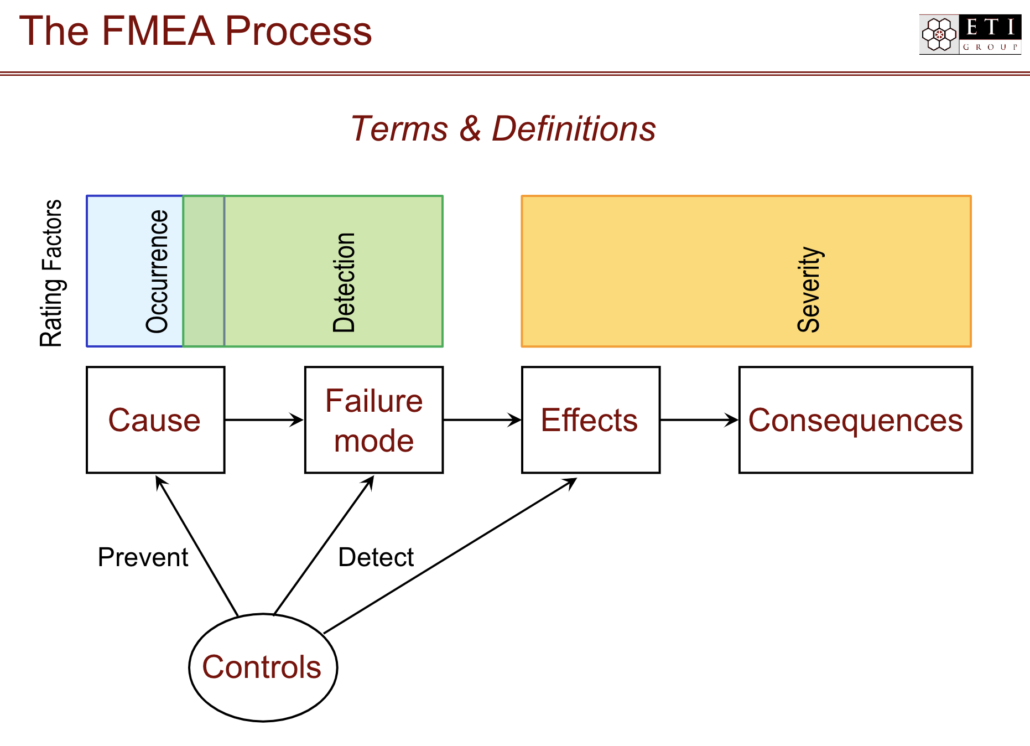
What is FMEA (Failure Mode and Effects Analysis)?
Failure Mode and Effects Analysis (FMEA) is a structured process for determining potential risks and failures of a product or process during the development phase. FMEA teams examine failure modes—those potential points of failure in a product or process — and what the failure effects (risks, harm, waste, or defects) might be for the purpose of mitigating or eliminating these potential failures before release.
Product development teams have to balance many competing interests. Market competition, customer demands, and stakeholder pressures can combine to tempt teams to rush products to market and skip important steps. But the risk of skipping steps also increases the risk of future failures, recalls, or legal challenges. How can development teams design and develop products that meet safety and regulatory guidelines, satisfy customers, and produce profits for the company?
One vital piece of the development team’s process is Failure Mode and Effects Analysis (FMEA). But in order to make the best use of FMEA, teams need to understand more than just what it is; they also need to understand why it’s important and how to integrate it fully into the product development process.
Failure Mode and Effects Analysis Process Overview for Product Teams
FMEA is more than just a one-time event or a prescriptive process meant to be checked off a list. Properly performed, FMEA can improve product quality, reduce costs, and help establish compliance with safety guidelines by helping development teams identify potential failures early in the lifecycle rather during verification or validation (or even post-market!). Performing FMEA also helps capture organizational knowledge around products and preserves information for future development cycles. Finally, collecting data on a new product not only documents what could go wrong with the product, but also what suggestions were rejected, what actions were taken, and what solutions were adopted.
It can be helpful to think of Failure Mode and Effects Analysis as highly structured brainstorming. Within the confines of an FMEA structure, team members are encouraged to consider all possibilities for product failure and use team and organizational knowledge to arrive at possible solutions or actions to solve the problems. Instead of being just a template to fill in, proper FMEA is a systematic group of activities intended to:
Recognize and evaluate potential failures of a product or process;
Evaluate the effects of failures;
Identify actions which could eliminate or reduce the chance of potential failure; and
Document the process and record decisions.
In this overview, we will review the history and purpose of FMEA, define key terms, and provide an outline for how to perform FMEA as part of the development process.
What is FMEA?
First used in the 1940s, FMEA started as a military process designed to test mission-critical safety. Since then, FMEA has expanded into industries as diverse as automotive, medical devices, and aerospace. As a tool to help mitigate risk and comply with international standards such as ISO 14971, ISO 13485, IATF 16949, and AS9100, FMEA supports many risk management tasks and helps establish proof of compliance.
What is a failure mode?
Failure modes are ways that an element, component, system, function, or process can fail. For example, a bicycle hand brake works by creating friction between the rotor and the brake pads. Events that interfere with that friction are failure modes — for instance, heavy rains might cause a reduction in friction when the brake is applied leading to failure of the brakes.
Once you know the failure mode, the effects can be determined. Effects are the ways that failures can cause harm, waste, or defects for the customer. In our bicycle example above, failure of brakes could lead to serious injury of the bicycle operator. The analysis piece of FMEA is where failure modes and effects are identified and prioritized so that they can be reduced or eliminated before new product release.
Failure modes are discovered through testing and brainstorming as part of the development process.
How is FMEA calculated?
What is a risk priority number (RPN), and how is it used in FMEA?
A risk priority number (RPN) is a number assigned to the specific process or step within a process to quantify the likelihood of detection or probability of occurrence of a failure mode and the severity of its impact. The RPN is the product of three scores: 1) the likelihood of the failure occurring, 2) the likelihood that the failure will not be detected, and 3) the impact of the failure in terms of harm or damage to a person or equipment.
In FMEA, every failure mode is assigned an RPN. The goal of the FMEA is to reduce the RPN; the higher the risk of severe impact, the more the FMEA process will seek to reduce it before product release or re-release through corrective actions.
FMEA can also simply be calculated by a simple SxO – severity times occurrence.
There are two types of FMEA that are most common: design FMEA and process FMEA.
Design FMEA
In a design FMEA (dFMEA), the product team reviews potential product malfunction, the possibility of reduced product life, and safety and regulatory concerns. The team looks at every aspect of the product, such as material properties, geometry, tolerances, interfaces with other components or systems, environments, user profile, degradation, and systems interactions.
Process FMEA
A process FMEA (pFMEA) looks for possible failures that reduce product quality or reliability and result in customer dissatisfaction. It also looks at safety concerns that might arise from human factors, processing methods, materials, machines, measurement systems, and environmental factors.
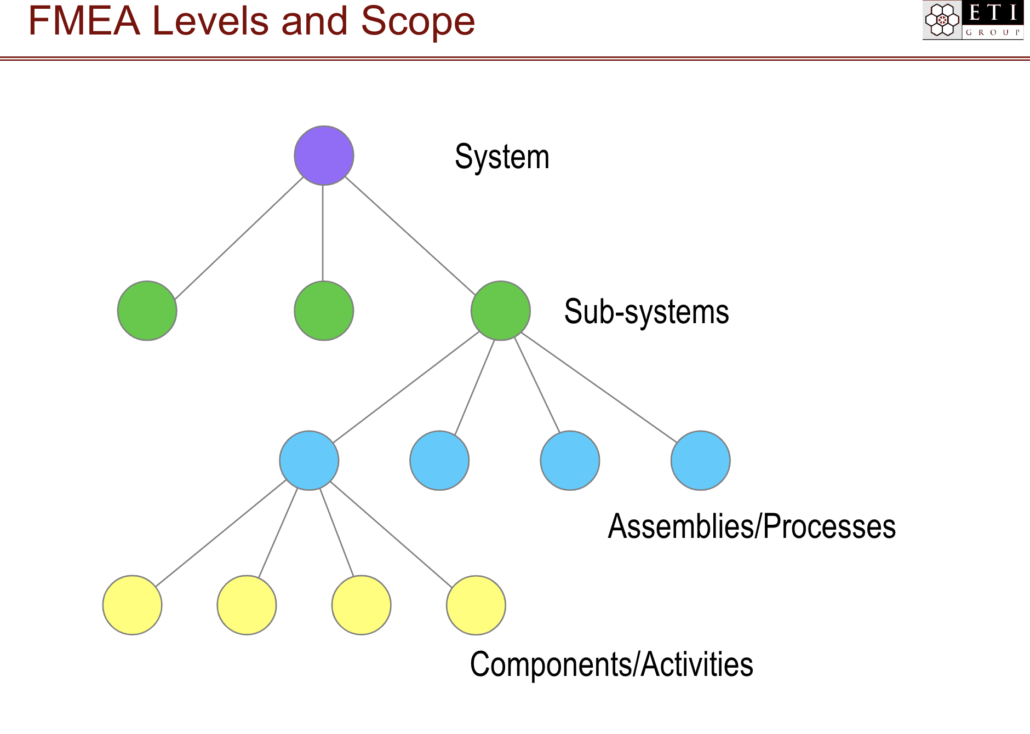
FMEA Levels and Scope
FMEA is context dependent; a proper analysis can only be performed if the team understands where the process or design feature lives in the overall system.
At the highest level, a product as a whole is one system. Under that system are sub-systems. Each sub-system has assemblies and/or processes, and each assembly or process has components and/or activities.
Image Courtesy of ETI Group 2019
FMEA should be performed at each of these levels and for each item within that level.
Within each item or level, the FMEA team needs to set the boundary of analysis, or scope. A clear scope will answer the following questions:
What is our focus—design or debug? (Where is the product on the development-to-launch timeline?)
Which item and level is being covered by this FMEA—system, sub-system, component, process, etc.?
What triggered this FMEA—changed regulation? Product redesign? New environment?
What aspect of design or process are we addressing (reliability, environmental impact, serviceability, etc.)?
Who is the customer, and what are the customer requirements?
For many companies and product development teams, the biggest worry around product release is a major misstep that results in harm to users, bad publicity, recalls, product corrections, lawsuits, or worse. But even smaller product issues can erode a company’s reputation and bottom line. If engineers and customer service teams spend their time fighting product fires and resolving customer complaints, they aren’t spending time designing new features and products to drive company growth.
Image Courtesy of ETI Group 2019
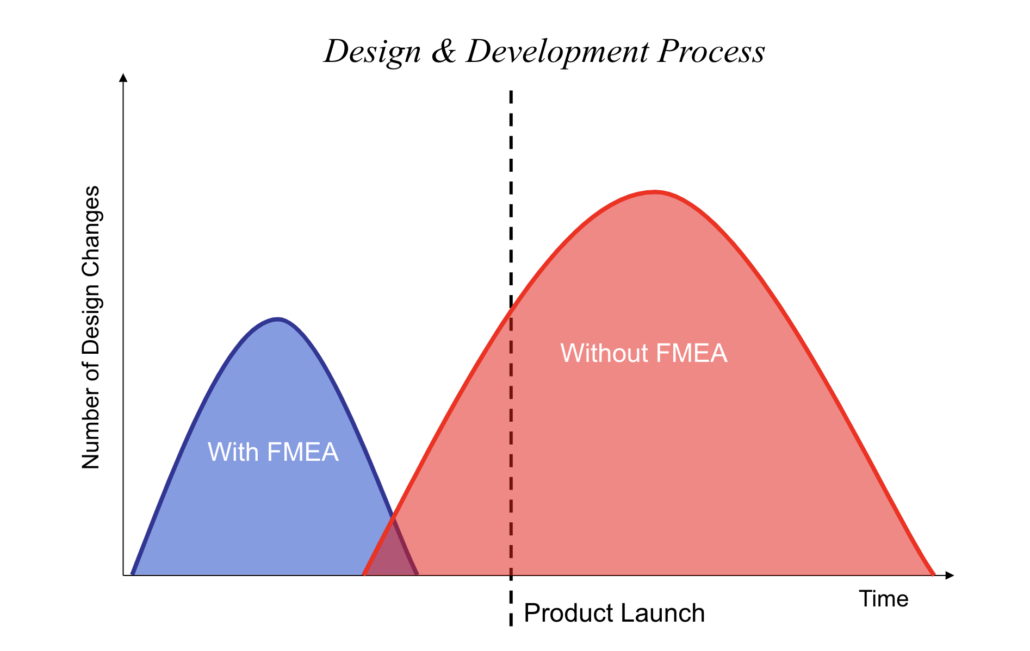
Making an FMEA procedure a routine piece of the product development process moves risk assessment to an early part of the development cycle. It helps ensure that risks are reduced early in the process, saving time, money, and reputation in the long run. Performing FMEA early in the process allows teams to think critically about potential design and process failures when it’s still possible to control risks, costs, and reputation.
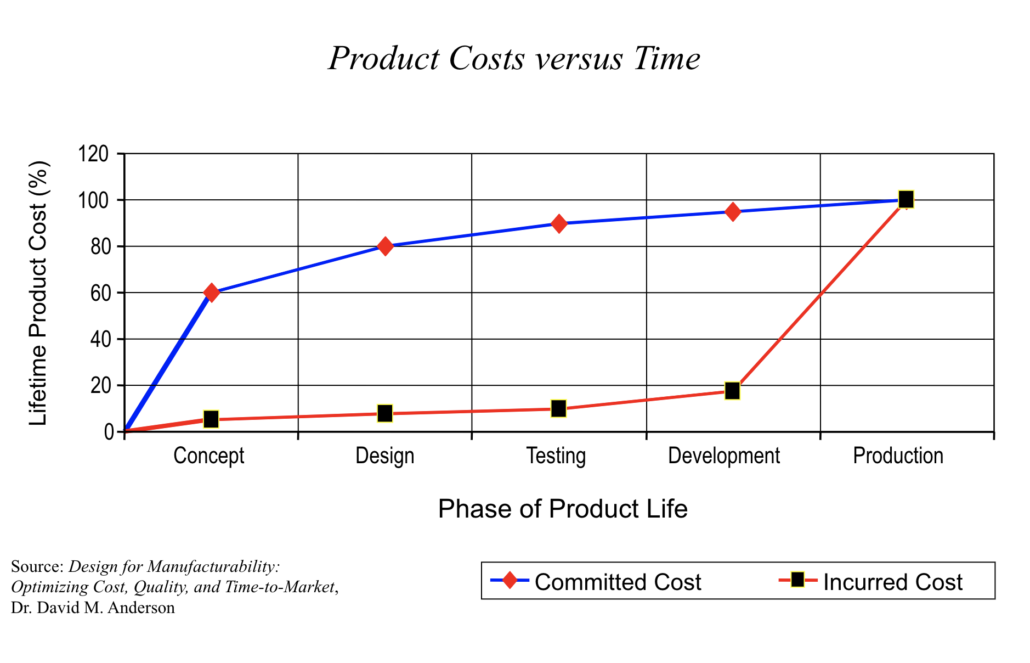
The time to control costs is early in the development process, before costs are incurred. Failure Modes and Effects Analysis helps companies accurately evaluate and plan for costs by providing a structured way to evaluate and analyze potential risks and failures.
Product cost vs. time graphic—Dr. David M. Anderson, Design for Manufacturability: Optimizing Cost, Quality, and Time-to-Market
When should we perform a FMEA?
FMEA is a proactive tool to use any time you are:
Developing a brand new design, technology, or process;
Modifying an existing design or process;
Changing the environment, location, application, or usage profile of a design or process; or
Responding to a change in regulation affecting a design or process.
It’s also important to remember that FMEA is a continuous process, not a one-time event. In fact, it is most effective when it is implemented at different times for different objectives across the development cycle. For best results, gather as many cross-functional minds as possible. This is not a call to have everyone in the company involved at every stage, but rather to gather people with different perspectives and insights who can focus on one system or sub-system at a time.
What is the FMEA process? How should we perform an FMEA?
While FMEA can seem overly structured at first, in reality, this structure is actually one of its valuable strengths. Within the FMEA structure, teams have the ability to ask questions, provide insights, and brainstorm solutions in a way that captures knowledge for the organization and improves product performance and safety at the same time.
But while the structure is a great strength of FMEA, it can be daunting for teams that have little experience with it.
Image Courtesy of ETI Group 2019
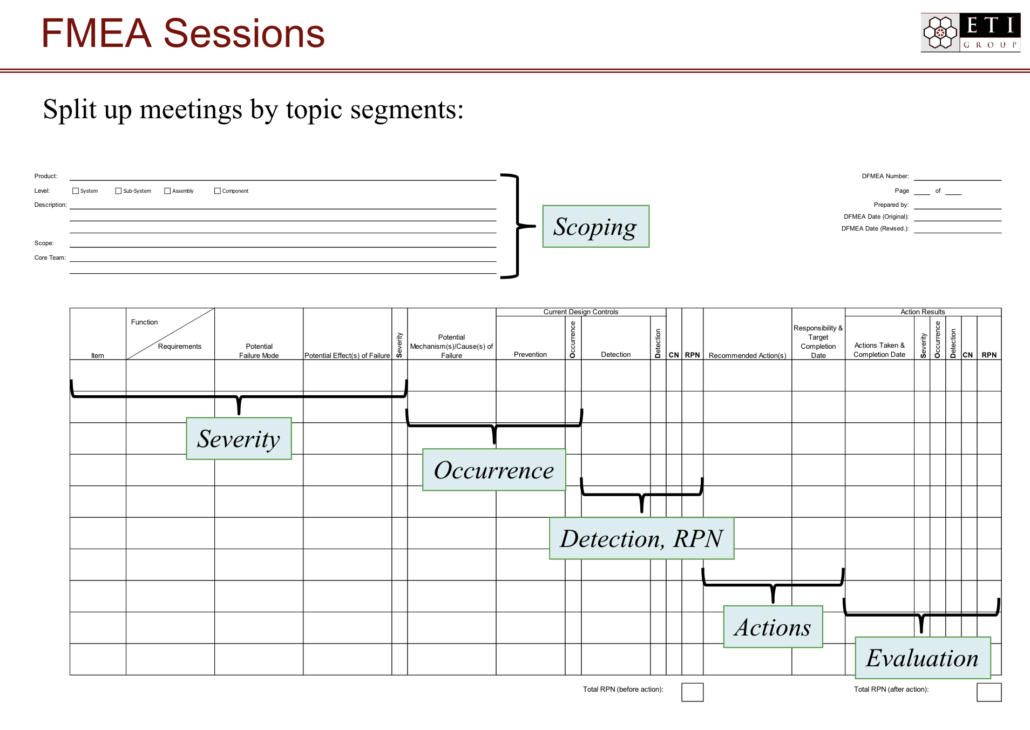
Here is an orderly way to approach FMEA:
Step 1: Prepare the FMEA document. Typically, this step can be done by a single person familiar with the design or process being analyzed—a member of the design team, for example. The temptation is to make this document too large or too encompassing. Remember, it should include only one system or sub-system or even one process or attribute at a time.
Step 2: Invite the team. This step can happen concurrently with step one. A member of the product team should form a non-permanent team of four to six people from inside and outside the development team. Your cross-functional team might include people from procurement, planning, finance, sales, marketing, and production—and, of course, the customer or end user.
Step 3: Input information. When the team is assembled and the document is in place, it’s time to brainstorm. Look at all potential failure modes. Evaluate potential causes, risks, and potential effects of the failure mode, and fill them in accordingly on the FMEA document.
Step 4: Assign RPNs. Calculating and assigning RPNs can often happen concurrently with inputting information, but if it doesn’t, be sure that these are assigned before moving on to prioritizing and brainstorming solutions.
Step 5: Prioritize failure modes. The failure modes with the highest RPNs are the most high-risk failure modes and should be evaluated and reviewed first.
Step 6: Coordinate with other teams. Of course, it’s preferable to have many FMEA teams reviewing systems, sub-systems, and other items within each level at the same time. Teams should be sure to coordinate with each other. If one team’s solution actually causes another team’s failure mode, or if one team’s effect could mean a consequence for another team’s system, it’s important to discover that as early in the development cycle as possible.
Step 7: Integrate with requirements management and compliance and regulatory guidance. As FMEA teams complete their analyses and resolve potential failures, those results should be integrated into the requirements management process and relevant safety and compliance guidelines. Using a requirements management tool such as Jama Connect, teams are able to link FMEA items directly to sources of potential failure and to mitigating requirements or verifications.
Image courtesy of ETI Group 2019
Are there templates for running an FMEA?
There are many templates available for running an FMEA. A simple search can turn up multiple results.
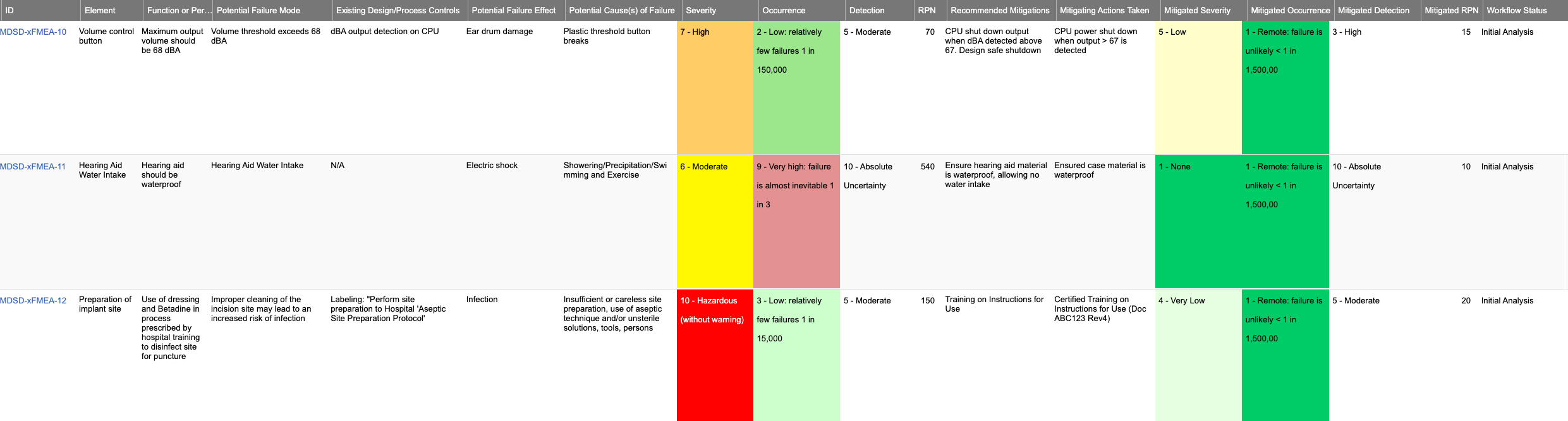
Heres a sample FMEA in Jama Connect
Are there software tools that can help streamline the FMEA process for product teams?
Jama Software integrates FMEAs directly into the design process by providing customizable templates that allow teams to collaborate, relate mitigations, track changes, review, and track workflow status.
Don’t let a rushed development cycle put you at risk for bad reviews, recalls, lawsuits, or worse. By integrating a thorough FMEA into your development process, you’ll bring better products to market and ensure safety and compliance.
To learn more about how Jama Software can help you connect your FMEA to your requirements management, contact us.
https://www.jamasoftware.com/media/2021/04/2021-04-28-fmea_1024x512.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2021-04-29 20:13:182023-01-12 16:49:16What is FMEA? Failure Mode and Effects Analysis Process Overview
Risk management plays a pivotal role in medical device development, helping ensure patient safety and product quality. Risk management is often misunderstood. It’s a process that could be cumbersome for organizations who don’t know where to get started, or worse, start too late in their development process.
After our recent half-day risk management seminar in the Netherlands, we sat down with award-winning medical device risk management educator, author, and consultant Bijan Elahi to hear his thoughts on risk management in medical device development.
Read the full interview below for Elahi’s take on the importance of risk management, best practices, and how use it to gain confidence in your compliance.
Jama Software: Thank you for joining us Bijan. For our readers who may not be familiar with your work, can you tell us a little bit about your background and experience with risk management?
Bijan Elahi: I’ve been doing systems engineering and risk management for the last 30 years, beginning my work in the aerospace industry at various companies, with my last job at NASA. In the early 1990s I was approached by the medical device industry for help with risk management. This was a time when there were no standards for medical device risk management, and medical device companies had no consistent way of doing risk management. I began helping one medical device company, then another, and soon I changed industries altogether, from aerospace to medical technologies.
The first version of ISO 14971 was published in the year 2000, finally providing risk management guidance to the global medical device community. I’ve been helping medtech companies understand that standard and apply it to product development ever since. In the last five or six years, I’ve also started teaching and consulting in risk management more broadly.
Jama Software: Looking back at your work in risk management, can you summarize for us what it is and why it’s so important to medical device development?
Bijan Elahi: Risk management, in a nutshell, is the process of identifying hazards, estimating the risks, controlling the risks, and then monitoring and reporting how you did. But once you identify the hazards, you also need to know why they happened, what are the causes of those hazards, and then what you can do to minimize the risk of harm from those hazards. Risk management is both an art and a science. It is not like mathematics that is perfectly clear. There are a lot of grey areas in risk management that require sound judgement and both a creative and a scientific approach.
Risk management is important because you can’t commercialize a medical product without it. It’s legally required to perform risk management before you can get approval to sell your product globally. Second, it helps you make safer products. We have a moral obligation to our patients to make the safest possible products for them.
It’s also important for business and financial reasons. Poor risk management can be expensive and often results in recall costs and even punitive damages. If you make safe products then you’ll have a better reputation, and you can sell more. In addition, it helps save money in product development, because risk management allows for the early identification of design weaknesses and allows targeted allocation of limited engineering resources with priority to the safety critical areas.
Jama Software: What are some risk management best practices product developers should consider as they’re moving through the development cycle?
Bijan Elahi: One of the things that medical device developers should recognize is that risk management should be done as early as possible. Even as early as the concept stage. Another thing is having a robust process for risk management that is both compliant and efficient. You can get really complex with risk management. So, avoid overcomplexity and have a process that is both understandable and explainable. Understandable to your own staff and also to regulatory bodies. When you have a regulatory auditor or reviewer asking questions about your risk management process, if you can’t help them understand, your process isn’t working. Because ultimately, you need to persuade a regulatory authority to approve your product.
Read our eBook to learn more about risk management for Class II and Class III medical device development.
Jama Software: You spoke in your presentation about an old vs. new way of thinking about risk management, can you expand on what you meant for our readers?
Bijan Elahi: The old way of thinking was where product developers saw risk management as a necessary evil. They would be focused on the design of the product and making it work. And then when they finished the design, they would say, “Well, if we’re going to commercialize this product, we need to show that we did risk management, and find somebody to do a retrospective analysis and write a report that this product is adequately safe.” That’s old thinking. The new way of thinking is to consider risk management as a value-added activity and a partner with product design. Risk management and product design should work together in lockstep. This way, we get real value from risk management.
In fact, regulatory bodies today expect this new way of doing the work. They don’t want to see that you didn’t do any risk management during the development process and that at the end you just wrote a report to retrospectively cover your bases. This way of thinking is not only bad for your company, it’s also bad for the consumer and bad for the product. A lot of times if you have finished your design, and then suddenly you make a discovery of a safety hazard, it may be too late to fix it or maybe just too expensive. Some companies would just try to somehow get the product out anyway, which is bad for everybody and doesn’t really save you any money.
Jama Software: Let’s talk a little bit about the latest version of ISO 14971 that was released at the end of last year. Have you had a chance to review the latest 2019 version and are there any new changes developers should be aware of?
Bijan Elahi: Yes, I have. ISO 14971 is the international standard for medical device risk management, and it is recognized by most countries. It is a very important standard because conformance to it is the easiest way to establish the safety of your medical device and to persuade a regulatory body to approve your device. The 2019 version has some changes in it, but they are not so extensive that they would overburden the manufacturers. There are some new definitions and some changes to existing definitions.
Also, there are some new concepts that are introduced. For example, in the 2012 version, the requirement was to reduce the risks “as far as possible.” In 2019, a new concept is introduced to reduce the risks to “as low as reasonably achievable.” And then in the previous version, 2007, there was another concept called “as low as reasonably practicable.” So, there are now three concepts in the most recent version about how can you strategize about reducing risks.
Jama Software: Speaking of changing industry regulations, one topic that’s been top of mind for medical device developers in Europe is the upcoming EU Medical Device Regulation (MDR). Is there anything that developers should consider when getting ready for MDR?
Bijan Elahi: Yes. MDR really created a ground shift for the medical device manufacturer. It caused a lot of change, and it’s more of a quality regulation. Some of the biggest changes with respect to risk management are the requirement to submit an annual Periodic Safety Update Report (PSUR). The PSUR is for Class II, and above medical devices. Another requirement is Post-Market Surveillance Reports (PMSR) for Class I devices. There will also be more emphasis placed on post-market clinical follows ups. The regulators expect you to continue to follow up your product and to see how it is clinically performing.
MDR puts more emphasis on post-market surveillance plans. You’ll need a plan ahead of time for surveilling your medical device and how it performs in the field. You’ll also need to identify and declare the lifetime of the medical device and provide a rationale. Article 88 in the MDR also requires trending, which will result in more reporting required by the manufacturer. If an adverse event hasn’t happened yet, but looks like it’s going to happen, then you have to report that too.
Jama Software: Interesting. So, you need to anticipate risks before something happens and there will be more documentation required?
Bijan Elahi: Yes. Before, we had vigilance reporting. Which means something bad has already happened, and you had to report it to the governmental agencies in Europe. Now in addition to vigilance, you have to predict if something bad is going to happen and report that as well.
Jama Software: Do you foresee any changes to risk management or requirements management being impacted by MDR?
Bijan Elahi: Good requirements management is just so smart to do for better and more efficient product development. I use it in risk management as well.
Basically, I treat hazards, risk controls, and safety requirements all as elements in the requirements management system. So, they’re all connected and traced. Traceability is another thing that is really emphasized in ISO 14971. It is so easy to do traceability with a requirements management solution like Jama software and, so hard to do it manually.
If you try to do it by manually, e.g., with an Excel spreadsheet, it’s so easy to lose traceability and have errors. It’s also hard to maintain. Anybody who has done traceability analysis knows how much work it is and how hard it is to maintain it because designs are dynamic. Things change all the time. Anytime you make a change, you could break your traceability. This is one thing that I always advise my students and my clients, to use a tool like Jama Connect.
Jama Software: Are there any specific best practices or any highlights that you can address in regard to risk and requirements management together? In regard to the discipline of combining those two and how they can help product development and launching products.
Bijan Elahi: Yes, I am a strong believer that risk-management and requirements-management are better together. For example, safety requirements: How do you know which requirements are safety requirements? A safety requirement is that which is linked to a risk control. As risk controls themselves are elements in the database, if you link some of your requirements to those risk controls then you can tag those requirements as safety requirements.
Sometimes when you are audited, an auditor asks you, “What are your safety requirements?” With the explanation that I just gave, it is very easy to run a query with a solution like Jama Connect for all of the requirements that are tagged as safety and get a list of them. Very easy to do! Without a tool, it’s not so easy to answer that question.
Another example is connecting Failure Modes and Effects Analysis (FMEA) analysis to your risk management. The tool can help manage the connections of the causes and effects that lead to hazards. Again, those causal factors themselves could be elements in a database which are linked together and that creates what’s called a sequence of events, which explains how a hazard can manifest itself. By creating this chain of events, you can connect the hazard to the hazardous situation, and subsequently to the harms. You can do a quantitative computation of risk, if you follow this methodology.
Jama Software: What are some common mistakes that you’re seeing medical device developers make in their risk management process?
Bijan Elahi: The first mistake that comes to mind is actually just an approach that is suboptimal. One of the things that many device manufacturers do when it comes to assignment of severity of harm is assign just one classification to it. For example, if you are infected, they would ask, “What’s the severity of the infection?” And they would assign a severity classification like serious or critical. These are key words that I’m using, by the way.
The thing is that a harm doesn’t create just one kind of outcome, but multiple outcomes. For example, an infection could cause anything from just a little bit of a fever, to organ failure, to even death. So, it’s not best practice to assign only one severity class to a harm. It’s best to assign a spectrum of severity classes to harms.
Jama Software: As we enter a new decade and medical devices are becoming more complex and more embedded in our everyday lives, how do you think risk management might change?
Bijan Elahi: Well, I think risk management is going to become more and more important. Especially with the introduction of EU MDR. EU MDR puts more emphasis on risk management. I see a lot more ‘risk-based’ language everywhere. Decisions need to be risk-based. That applies to all kinds of decisions, including design changes and verification testing. If you’re going to make ‘risk-based’ decisions, you need to know what risk is. And where you get risk information: is from risk management. Risk management is a progressively more prominent endeavor in medical device development.
https://www.jamasoftware.com/media/2020/02/Risk_Management_Blog.png5121024Bijan Elahi/media/jama-logo-primary.svgBijan Elahi2020-02-13 04:30:512023-01-12 16:50:45An Interview with Award-Winning Medical Device Risk Management Educator and Author Bijan Elahi
You’ll gain more confidence that your recently-released products avoid recalls, fines, or worse if you perform failure mode and effects analysis (FMEA) as part of your risk management.
Over the course of the hour-long webinar, Quillinan and Jama’s senior product manager, Joel Hutchinson, provided some key elements around the value of risk management and FMEA, and the importance of modernizing your risk management process to accommodate.
At the conclusion of the webinar’s presentation, the pair answered a range of questions from the hundreds of participants in attendance. Unfortunately, they weren’t able to get to everyone’s questions, but luckily they took some extra time afterward to answer some of those remaining, touching on everything from risk mitigation in aerospace to specific standards like ISO 14971.
We’ve compiled the questions (some of which have been slightly modified for clarity) below and encourage you to check out the full webinar and Q&A session here.
Q: Given that FMEA teams in companies can be non-permanent and have a fluid scope, which role should own and drive the activity overall?
Bethany Quillinan: I think the ownership role should align with those of the design/new product introduction process. If there is a project lead, then that person would make the most sense. There may also be a particular functional group who is given ownership of risk management, like Quality or Compliance teams. I think it really depends on the organization. What we want is alignment and integration within the design process, not a separate “silo” for risk management.
Q: Do you have any suggestions for criteria that would help when selecting a good FMEA facilitator?
Quillinan: A good facilitator will be someone who is well-versed in the FMEA process and the organization’s particular methodology, rating scales, etc. Additionally, the facilitator should be skilled in well, facilitation, and by that I mean having meeting management skills — things like tracking time, noticing when energy is low and a break is needed, drawing out quieter members, dealing with overly-dominant members, and generally equalizing the field. The facilitator should not get involved in the “content” of the FMEA, just the process itself. Ideally, there is also a content leader who can keep the team focused on the scope of the FMEA. That said, the facilitator should be aware of the scope, in case the team gets way off track and no one is calling it.
Gain Confidence in Your Risk Management Plan with Jama Connect. Learn how.
Q: Any suggestions for facilitators to keep people from taking things personally?
Quillinan: One engineer I worked with who had facilitated a lot of FMEAs shared a good technique — to ask people, “How could you make it fail?” And that turned around defensiveness to creativity. From a facilitation aspect, I think emphasizing that the analysis is “potential,” that we’re not saying it’s going to happen, but that it’s a just a possibility to consider. Depersonalizing statements can help — talk about “the design” vs. “your design,” for example. If someone is super defensive, the facilitator may need to call a break to let things cool down and talk to the person offline. Diplomacy is important!
Q: What you talked about is used in the automotive field which is very effective, but the aerospace industry tends to use FMEAs focused on managing the effects and quantitative failure rates (assuming constant failure rates). How do you get the benefits of automotive FMEA and, at the same time, satisfy aerospace FMEA requirements?
Quillinan: Having quantitative failure rate data to inform the discussion of the probability of occurrence is a big benefit. Failure mode, effects and criticality analysis (FMECA), where “C” is criticality analysis brings in more of the quantitative factor, and I see that terminology used more in aerospace. Satisfying specific Aerospace requirements is somewhat out of the scope of this presentation. In the context of the AS9100 quality management system standard, the requirement is to perform risk management, and FMEA is not prescribed but is a possible tool. Bottom line, I see FMEA as a prioritization tool rather than a reliability prediction. (In the early days, the military hoped to use FMECA to calculate an actual reliability metric, but it didn’t work out that way.)
Learn why Frost & Sullivan likes Jama Connect as a modern solution for risk management. Read the brief.
Q: What happens when multiple users are creating individual risk analyses? How do they collaborate if they are analyzing similar things?
Joel Hutchinson: As Bethany mentioned during her presentation, we understand that there’s going to be overlap in various FMEAs. An example of this is subsystems, which may have additional functionality that is only present at the system level. Risk management in Jama Connect helps the cross-functional team that works together to perform a risk analysis and has the ability to add view-only roles for context. One way of addressing this would be for the moderators of the two overlapping risk analyses to add each other as view-only roles to their respective analyses. This way, each cross-functional team is aware of what the other team is doing and how they’ve scoped their risk analysis.
Do you know where, when, and how intended Use FMEA (uFMEA for medical devices, per IEC 62366-1:2015) and software FMEA (life cycle requirements for medical device software per IEC 62304:2006+A1:2016)integrate into the requirements management process and product development timeline?
Quillinan: Generally speaking, risk management for usability, software, or any other aspect of a design should be integrated with the design process as soon as these aspects enter the design conversation. For example, early on during the initial “concept” phase, we need to be thinking about usability risks to inform the design concept and assist in evaluating different design routes to take. I also think useability/human factors could potentially be considered at any level of the product, so it could follow the same general timeline I showed in the presentation.
Likewise, for software, at the concept phase, the conversation will likely include software needs for satisfying customer expectations. From a systems standpoint, I feel the sooner the better. (I often use the initial rollout of the Healthcare.gov website for the Affordable Care Act (ACA) marketplace as an example of a “silo’d” approach to design. Each module was developed and tested in isolation and the “validation” of interfaces between modules was when it went live… and crashed.
We have to remember that the ultimate focus is at the system level — the end-user interaction with the product. In a medical device setting, most consultants are going to advise you to build in human factors considerations throughout the design process rather than waiting until the final human factors validation test on the finished design, for all the same reasons I described in the presentation.
Find out how to better manage medical device development in accordance withISO 14971. Read our guide.
Q: Risk Priority Numbers (RPNs) seem to always be subjective from one party to another, specifically the occurrence and detection. The occurrence is difficult to gauge when the failure mode and risks are new or when little-to-no history is available. For detection, we’ve had difficulty assessing the controls in place and how to rate them in terms of efficiency.
Quillinan: One way to help make the RPNs more objective is to establish and use consistent rating scales with descriptions that are relevant to your organization’s products and processes.
When there is no history and risks are new, the typical practice is to be conservative and give them high ratings. I often find that people have difficulty distinguishing prevention controls from detection controls.
Preventive controls typically act on the inputs to a process or are the controls during the process — think of mistake-proofing, for example not being able to choose an obsolete revision of a component for a bill of material (BOM) or having design guidelines to follow. Detection controls are after the fact and are inspections of the output of a process. In design, a typical detection control is a second person (or more) performing a design review.
Q: What is the relationship between design failure mode and effect analysis (DFMEA) and process failure mode and effect analysis (PFMEA), in terms of the failure modes and causes?
Quillinan: In DFMEA, we are looking at how the product could fail, and causes are from the design process itself; the assumption is that the process is being run correctly. In PFMEA, we’re looking at how the process could fail, assuming the product is designed correctly. Of course, this depends on FMEA scope. If we’re looking at a design for manufacturability, then we’re looking at how the design process could fail in terms of designing the product in a way that it can be easily and efficiently built.
https://www.jamasoftware.com/media/2019/07/Frost_and_Sullivan_Regulated_Industries.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2019-07-23 04:00:402023-01-12 16:51:32Answers to Your Questions About Risk Management and FMEA
No one sets out to create a product that’s an abject failure, which is one of the reasons Failure Mode and Effects Analysis (FMEA) exists.
FMEA is one way to identify the many ways something could go wrong with, among other things, a product or service. Understanding how a product could malfunction and what can be done to prevent it is an especially important task in safety-critical markets such as aviation, automotive, and medical devices.
Even still, development teams are under such tight deadlines to bring products to market quickly that approaches like FMEA can get brushed aside. Then, when a problem with an already-released product emerges, there’s a mad dash to correct things, which can throw an organization’s momentum, strategies, and resources off course.
“Do you want to think about this stuff early and ahead of time, when it’s private and less expensive?,” asks Bethany Quillinan, Senior Quality Systems Consultant with Oregon Bioscience Association, “Or, do you want to wait until it’s public, embarrassing, and you’ve had a big recall, or you’re testifying in front of Congress?”
What is FMEA?
Failure Mode and Effects Analysis (FMEA) originally started in the military in the 1940s, as a way to test mission-critical safety. It’s since expanded into a variety of industries, perhaps most notably in the automotive industry.
It’s mainly used for risk management with international standards like ISO 14971 and ISO 13485 (medical devices), IATF 16949 (automotive), and AS9100 (aerospace). In these types of regulated industries, FMEA is a way to accomplish a lot of risk management tasks to provide proof of compliance.
FMEA’s value extends far beyond the simple checking of a box for compliances purposes though. In performing FMEA, you’re not only capturing organizational knowledge around a given product, but you’re also simultaneously preserving that information for future development cycles. And by collecting that data, you’re including not only what could go wrong with a product, but also the decisions that were made, actions that were taken, and suggestions that were rejected.
“It preserves history for people,” Quillinan says. “It’s that hackneyed saying of, ‘If you don’t remember your history, you’re doomed to repeat it.’ We don’t want to do that in product development.”
Why Use FMEA?
One of the biggest mistakes a company can make around risk is thinking you’re infallible from a gigantic misstep. And when something like that does happen, it can be a major shock, complete with bad publicity, recalls, product corrections, lawsuits, and worse.
Even if those heavy consequences don’t come to bear, even light issues with your product can eat at your reputation and bottom line, sending everyone from engineers to your customer service team running backward and extinguishing fires. And if your teams are constantly running around firefighting, they’re not spending that time designing new features or pushing your product forward.
“So often, we’re reworking things, we’re rewriting things, we’re doing failure analysis, we’re appeasing unhappy customers, we’re fixing bugs,” Quillinan says. “From that day-to-day level, it’s a huge energy, time, and money drain of dealing with stuff after the fact.”
Plus, when it comes time to develop the next generation of product, you’ll likely already be behind on time and, odds are, the competition.
When to do FMEA?
It’d be hard to imagine a company releasing a product after a single design review.
In a similar way, FMEA is not a one-time endeavor limited to just the risk management process. FMEA should be done continuously throughout the development cycle and be used for different objectives at different points.
Unfortunately, many developers look at FMEA initially, realize it’s going to be a lot of effort, and try and do it all at once. And that can become overwhelming for everyone involved, further limiting the work to only those who are involved in the risk management process.
To ensure the best results with FMEA and risk analysis, it helps to have as many cross-functional minds thinking about what could go wrong with your product as soon as possible.
https://www.jamasoftware.com/media/2019/07/Product_Requirements_Document_Header_Image.png5121024Jama Software/media/jama-logo-primary.svgJama Software2019-07-02 04:00:392023-01-12 16:51:34Why You Need to Make More Time for FMEA
While a positive brand reputation takes years to nurture and build, irrevocable damage to a brand — and a business — can happen in only moments.
As innovative companies are asked to move faster than ever to bring products to market, it’s worth taking a moment to step back and look at the cost — financial and otherwise — of a misstep that could lead to product recall or failure. In addition to fines, regulatory reprimands, and decreased market share, a product recall inevitably impacts your brand and can have a major impact on your bottom line.
Brand erosion, for instance, is one consequence of a major product recall. As the name suggests, brand erosion is the gradual destruction or diminution of your organization’s reputation when your products are deemed unsafe, unfit for the marketplace, or simply don’t perform as marketed.
If you’re working in an industry where functional safety is a top priority, like automotive or aerospace, guarding against these types of scenarios means getting crystal clear on the standards and regulations that help protect your product. That also means ensuring proper risk management techniques like Preliminary Hazard Analysis (PHA) and failure modes and effects analysis (FMEA) are being performed correctly.
Often times, penalties come as a result of companies not properly performing these functions, and instead rushing to deliver products that haven’t met benchmarks for quality, compliance, and — in the case of many industries — functional safety. Other times, companies simply fudge the data. Let’s look at a few famous examples:
Ford Motors’ 1980 Failure to Park Product Recall
While product recalls can happen in any industry, some of the most notorious and detrimental have happened to automotive companies. Such was the case for Ford Motors which, in 1980, was determined by the Department of Transportation to have more than 23 million cars and light trucks in circulation that contained a functional safety defect that permitted the vehicle to accidentally slip from park into reverse, although to the driver the placing of the shift lever appeared to be in park.
And while the company would have faced financial ruin if all 23 million automobiles had been recalled, regulatory authorities ruled that instead of recalling all vehicles involved, Ford could make amends by sending out a sticker for drivers to place on their dashboard warning that “unexpected and possibly sudden vehicle movement may occur” if the car was not properly parked.
The failed safety catch was found to have led to 6,000 accidents, 1,700 injuries, and 98 deaths, and Ford was tied up in litigation for years, costing the company tens of millions of dollars. The “failure to park” issues continued even after the sticker warning was issued.
While the cost of this functional safety error included lost revenue and expensive lawsuits, the safety malfunction may have additionally cost Ford the trust of their consumers. Especially in industries that are highly regulated — and thus highly trusted — recalls related to safety and reliability can cause devastating harm to the foundation of the organization.
Learn how how a Fortune 100 semiconductor company is meeting the challenges of developing automotive-related technology by downloading our case study.
Volkswagen’s “Defeat Device” Scandal
While some product failures and recalls are the result of honest mistakes, such as the example above, others can be attributed to downright dishonesty. Such was the case for Volkswagen in September of 2015, when the company admitted that they had installed software in 550,000 automobiles that could figure out when the cars’ emissions were being tested and modify their performance to meet mandated standards.
Initially, Volkswagen spokespeople claimed that a “few software engineers” were struggling to meet stringent U.S. emissions standards while also producing a diesel engine that would perform well. That, the spokesperson said, was the reason those individuals decided to design a “defeat device,” a system to switch on emissions controls when the cars were being tested and turn them off when the automobile was driving normally. What we now know is that knowledge of this system went much higher, and some believe awareness could have been as high as the CEO, who resigned and was later indicted.
The scandal cost the company over $29 billion dollars in recalled automobiles, but the damages didn’t stop there. After losing the trust of the public, the company posted their first quarterly loss in 15 years, showing that consumers weren’t likely to invest their money in an organization that had deceived them.
A recent study by Edelman shows that consumers are more interested than ever in purchasing from organizations that they believe are “doing the right thing.” The study revealed that 64% of consumers around the world — up 13% from 2017 — now buy on belief, meaning that they will choose, switch, avoid, or outright boycott a brand based on where the organization stands on issues they care about.
The same study showed that consumer trust dropped 10% (from 58% to 48%) in the last year, and that nearly half of consumers don’t trust businesses to “do what is right.” And organizations whose products are recalled due to deceiving functionality are certainly not going to earn a reputation for doing what’s right for their customers.
An additional 8 million cars were sold in the E.U. with the same “defeat device,” but because emissions standards are much lower there than in the U.S., Volkswagen legally committed no crime in the E.U.
NASA’s Costly Math Mistake
On December 11th, 1998, NASA launched a robotic space probe called the Mars Climate Orbiter into space with the hope of studying the Martian climate, atmosphere, and surface changes. Nearly 10 months later, in September 1999, the $125 million probe burst into flames and fell to pieces in outer space. While this was certainly not the first, or last, failed space expedition, what sets this one apart is that the reason for failure comes down to a simple math error.
The navigation team at the Jet Propulsion Laboratory (JPL) used the metric system of millimeters and meters in its calculations, while the team that designed and built the spacecraft, Lockheed Martin Astronautics in Denver, provided crucial acceleration data in the English system of inches, feet, and pounds.
The result was that the software controlling the orbiter’s thrusters was faulty. The software calculated the force that the thrusters needed to exert in pounds of force, while a second piece of code that read this data assumed it was in the metric unit— newtons per square meter. And while fortunately no lives were lost, this simple mistake pushed the orbiter dangerously close to the planet’s atmosphere, destroying the spaceship and killing the mission.
Despite NASA’s next Mars mission also resulting in a lost spaceship, the organization did bounce back from the ordeal. They recovered over the following years by returning to the basics, rebuilding the Mars program based on conservative strategies and concepts that had already been tried and tested.
In the case of NASA’s space mishap, detriment took the form of missed opportunity and lost ground in their research efforts. Because of the immense time, money, and resources it takes to launch a space mission, failures and missteps can result in years of setback. What was supposed to be the first weather observer in another world became a $125 million mistake.
How Better Requirements and Risk Management Can Help You Avoid Product Recalls and Failure
Developing complex systems and products requires teams to have the ability to effectively define and track requirements, adhere to safety-critical regulations, collaborate and communicate effectively across teams and functions, and evaluate and mitigate potential risks. Failure to do so may result in product recalls or failure, and in the case of safety-critical products, injury or worse.
According to the CHAOS Reports from The Standish Group, three of the biggest contributors to projects that fail are lack of user input, incomplete requirements and specifications, and changing requirements and specifications.
Unfortunately, too many organizations struggle with requirements and risk management and the effects, some outlined above, can be devastating.
In the case of the Mars Climate Orbiter, the simple miscommunication between JPL and Lockheed Martin not only shows how small mistakes can lead to mission failure, but also highlights the need for teams — especially those in different physical locations — to work collaboratively instead of in silos, and for organizations to invest in solutions that help teams achieve this.
With the Jama Connect Risk Management Center, development teams can participate in risk management techniques including PHA and FMEA in accordance with industry-specific standards like ISO 14971 and IEC 60812. By working with live data, teams can efficiently identify and mitigate risks early in the development process, ensuring quality and safety in complex product development.
To learn more about how Jama helps organizations thrive in critical product markets by reducing risk and providing a single source of truth, download Frost & Sullivan’s recent executive brief,“Safeguarding Regulated Products Amidst Growing Complexity.”
https://www.jamasoftware.com/media/2019/05/Brand_Erosion_Product_Recall.png5121024McKenzie Jonsson/media/jama-logo-primary.svgMcKenzie Jonsson2019-05-28 05:00:412023-01-12 16:51:37Avoid Product Recall and Failure with Better Requirements and Risk Management
 Incorporate Design Controls in Medical Device Product Development") Incorporating Design Controls can be a daunting task for medical device product development teams. For a variety of reasons, including unfamiliarity of regulatory expectations, inexperience with implementation, or viewing them as a burden preventing innovation, too many teams incorporate Design Controls reactively instead of proactively.
Incorporating Design Controls can be a daunting task for medical device product development teams. For a variety of reasons, including unfamiliarity of regulatory expectations, inexperience with implementation, or viewing them as a burden preventing innovation, too many teams incorporate Design Controls reactively instead of proactively.