In this blog, we preview our whitepaper “FDA New Draft Guidance of Premarket Submissions for Medical Devices: Are you ready?” Download the entire whitepaper to learn more!
FDA New Draft Guidance of Premarket Submissions for Medical Devices: Are you ready?
Technology innovation has undergone rapid transformation since the Food and Drug Administration first developed its “Guidance for the Content of Premarket Submissions of Software Contained in Medical Devices” in 2005. Over the past 17 years, significant advancements have disrupted the market, including the advent of the smartphone, watches that monitor your sleep and snoring habits, and smartphone-connected pacemaker devices, just to name a few.
The medical device industry has also undergone a sizable transformation, which is why the FDA recently released the “Premarket Submissions for Device Software Functions” draft guidance to keep up with the changes. Please note that while this guidance is valuable to medical device developers to get a sense of what’s to come, it is not currently being enforced.
It’s also important to note this guidance applies to all premarket approval (PMA) and to some 510k’s, based on risk level.
The new draft guidance, which as of March 2022 has not yet gone into effect and therefore is currently non-binding, revises documentation standards that apply to the software development, design verification, and design validation processes.
As you work to adapt to the new guidance, you might have many questions, including “Who and what does the new guidance apply to? And what exactly has changed?” Answering these important questions will assist you with getting ready for the changes, which are set to finalize in spring 2022.
The New Guidance: Does It Apply To Your Device?
The new guidance applies to all premarket submissions that include at least one device software function. However, it’s not intended to provide recommendations on how a device’s software should be developed, verified, and validated. Software in a medical device (SiMD) and software as a medical device (SaMD) are both included in the guidance. The following are also included, according to the FDA:
Firmware and other means for software-based control of medical devices
Stand-alone software applications
Software intended to be operated on general-purpose computing platforms
Dedicated hardware/software medical devices
Accessories to medical devices when those accessories contain or are composed of software
Recent changes made to section 520 of the Federal Food, Drug, and Cosmetic Act under the 21st Century Cures Act excludes some types of software, including those that are lower-risk, support software, such as certain mobile applications for consumers, and software for administrator support to laboratories. Additionally, the guidance does not apply to automated manufacturing and quality system software or software that is not a device.
To learn more, download the entire whitepaper where we cover a summary of the draft guidance, plus:
The difference between the new and old guidance
Documentation levels: Basic vs. Enhanced
Software documentation elements
RELATED
https://www.jamasoftware.com/media/2022/05/FDA-pre-draft-submissions-1.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2022-05-26 03:00:312023-01-12 16:46:40FDA New Draft Guidance of Premarket Submissions for Medical Devices: Are you ready?
One of the early steps I advise my clients to take when developing their medical device is to determine the class and classifications of their medical device. In conjunction with the complexity of the device, understanding the class and classification sets the foundation for your product development timeline and effort.
This post gives a basic introduction to FDA medical device classes and classifications and the implications for your product development schedule and requirements management.
What are FDA medical device classand classifications?
The FDA established three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. Classification is based on the intended use of the device and indications for use, as well as the risk the device poses to patients and users.
There are three classes: Class I, Class II, and Class III. Class I devices are those with the lowest risk, Class II devices have a greater risk, and Class III includes devices with the greatest risk.
The FDA also established classifications for over 1,700 generic types of medical devices and grouped them into 16 panels, or medical specialties. Example panels include Cardiovascular Devices and Radiology Devices. Each of the generic types is assigned as Class I, Class II, or Class III.
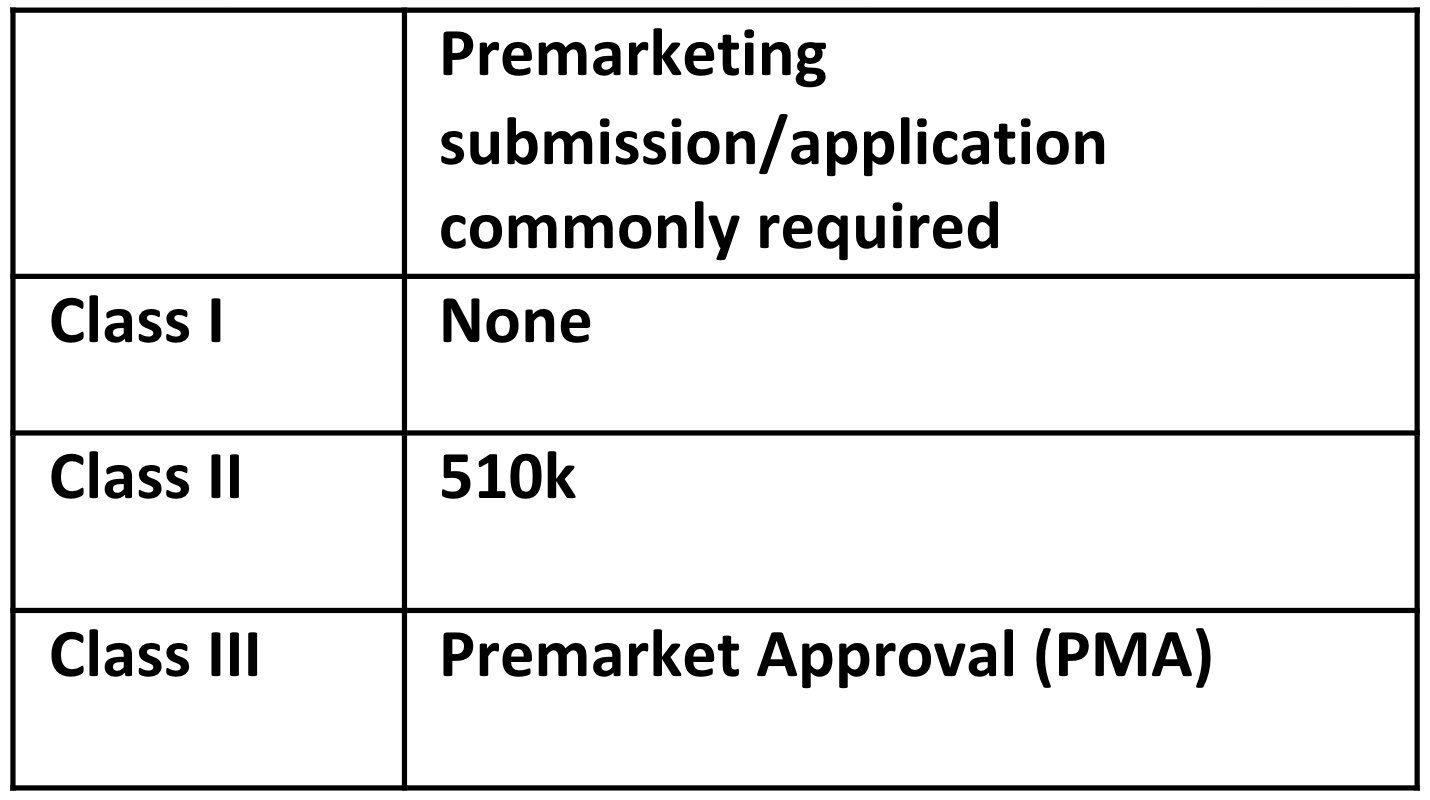
The class and classification of the device impacts what FDA premarket submission or application is required for clearance to market. The common premarketing submission or application for each class are:
Note: These are the common regulatory submission and applications for each class of device. There are exemptions, limitations on those exemptions, special controls that may apply, and exceptions, so be aware whether any of these applies to your device. For example, about a quarter of Class I devices are not exempt, and a 510k premarket submission is required.
As the process for the 510k submission is 30-90 days, and the process for the more in-depth PMA submission is 180 days to accept or reject, this time should be understood and planned into your product development schedule.
Similarly, expect elements from the required design control process and design history file to be included as part of a 510k and PMA. Also keep in mind that when design controls are required for your device classification, the full design history file can be scrutinized as part of an FDA inspection of your organization. Since the FDA evaluates whether a device is effective and ensures the risk to the patients and users is appropriately addressed, good requirements and risk management is key. It’s important to have an organized manner in which to demonstrate and document that risk management and user needs are successfully traced through design inputs, design verification, and design validation. A requirements management tool like Jama Connect™ allows for this traceability in an efficient, collaborative, and regulatory-compliant manner.
Understanding your device class and classification is a key step to understanding the path for FDA regulatory clearance and subsequent design control requirements for your medical device development. Knowing those expectations up front will make for a smoother medical device development journey.
Learn more about developing medical devices with Jama Connect!
https://www.jamasoftware.com/media/2022/01/2022-02-18-understanding-fda-medical-device.jpg5121024Michelle Wu/media/jama-logo-primary.svgMichelle Wu2022-02-18 03:00:282023-01-12 16:47:36Understanding FDA Medical Device Class and Classifications, and its Impact on Requirements Management
In many ways 2021 was a continuation of the changes brought about in 2020, a year that’s been described as “unprecedented” and “unparalleled.” In a unique way, 2021 has offered us an idea of evolving innovations and technology on the horizon for teams across industries. These changing conditions will present a variety of new landscapes and will offer unique challenges, opportunities, and more than likely, many surprises.
As we enter a new year of further changes, Jama Software asked select thought leaders – both internal and external – across various industries for the trends and events they foresee unfolding over the next year and beyond.
In the second part of our five-part series, we ask Steven Meadows, Solutions Lead from Jama Software, and Ryan Moore and Carleda Wade, Solution Consultants from Jama Software – along with Thierry Marchal, Program Director for Healthcare Solutions at Ansys, and Ivan Ma, Medical Device Program Leadership, to weigh in on product and systems development trends they’re anticipating for medical device teams in 2022.
Read our other 2022 Industry Predictions here: Part One – Engineering Predictions,Part Three – Automotive Predictions, Part Four – Aerospace & Defense Predictions, and Part Five – Insurance Development Market Predictions.
Medical Device Predictions
Q: What product, systems, and software development trends are you expecting to take shape in 2022?
Steven Meadows, Jama Software:
Artificial Intelligence (AI)
AI, and in particular machine learning (a subset of AI), has been expanding rapidly over the past decade and now has a market of over $6.7 billion across med tech. I see this trend continuing with extra guidance from the FDA helping developers produce safer software with AI elements. AI is particularly exciting as it can enable streamlined MRI and CT scans, instant blood and at-home rapid testing, and a whole host of otherwise manual and often error prone activities.
Minimally invasive devices
Another trend I see in medical continuing in 2022 and beyond is the rapid growth and development of minimally invasive devices. Open heart surgeries are becoming a thing of the past, with cardiothoracic applications becoming the norm. Minimally invasive devices are also becoming heavily utilized throughout most orthopedic surgeries, and increasingly in urology procedures. We have seen a lot of start-ups attempting to bring the next best minimally invasive device to market.
Wearables
Medical wearable devices demand continues to rise, with apps on wearable devices beginning to provide recommendations to users, to improve their health. Heart rate and temperature sensing continue to be the most popular features on wearable tech today but there is increased investment in smart glasses, ‘earables’, and clothing.
Software development
Software will continue to be an integral part of many medical devices next year and beyond. We consistently see that our software customers have adopted, or are transitioning to, an Agile methodology when creating their medical system, a shift that has been happening for some time. Gone are the days of slow waterfall-based development practices.
As I mentioned before, AI based software development will continue to trend and grow next year, and AI software development practices will be geared around a tailored regulatory framework, good machine learning practices, and a patient centered approach. The FDA will continue to refine guidance around AI/Machine Learning (ML.)
Cybersecurity and in particular data security will continue to be a top priority for our software customers, with data breaches and hacks on the rise. The FDA is planning to soon release extra guidance around cybersecurity in medical devices with a particular emphasis on quality system considerations and content of premarket submissions.
Ryan Moore, Jama Software:
I anticipate a trend toward more software-basedtoolsets and digitization. Also, an increased amount of automation and robotics.
Carleda Wade, Jama Software:
I expect greater integration of software tools such as with Requirements Management software with eQMS software.
Thierry Marchal, Ansys:
The dramatic COVID pandemic has amplified a trend that appeared a decade ago – progressively calling for the adoption and deployment of in silico medicine. Stormed with the COVID pandemic, the world could not wait for 10 to 15 years to get a new vaccine fully tested and approved using a traditional approach. Furthermore, this COVID disease is impacting people differently, while elderly people are impacted the most, possibly leading to long term disabilities and treatments.
EDITOR’S NOTE: According to The University of Sheffield’s Insigneo Institute, In silico medicine (also known as ‘computational medicine’) indicates modeling and simulation technologies that directly contribute to the prevention, diagnosis, prognosis, treatment planning & execution, or management of the disease. In silico methods complement traditional in vivo approaches (working with animals and human beings) and in vitro testing (working in a lab.)
Thierry Marchal, Ansys:
This pandemic highlights what we have been observing for decades: with an aging population, the cost of chronic diseases (especially for old people) is weighing a lot on the healthcare cost. The solution will come from personalized medicine and preventive medicine combining pathologies detection in its early stage to treat diseases before they impact people. The continuous monitoring of patients to detect pathologies early are calling for e-health and mobile health (m-health): wearable and implantable devices continuously and safely watching the patient and soon feeding your own digital twin or Personal Digital Avatar (a computer model of yourself, connected with you, and properly stored in the cloud to guarantee your data privacy) is now an emerging trend. Digital twins will be a crucial innovation for the software necessary to use patient specific data to predict the evolution of the connected patients.
As medical innovation will be essential soon, this evolution cannot be slowed down by an extremely long and costly regulatory approval process: a digitalization of drug and medical device approval, including in silico (clinical) trial is another major trend that we are observing.
Ivan Ma, Medical Device Program Lead:
Technologies that enable capabilities via telemedicine with more than just a face and a voice over the screen will make “seeing” the doctor digitally as common place as working from home.
Protecting patients, physicians, and staff by reducing or eliminating exposure to harmful forms of imaging such as fluoroscopy will always be a valuable endeavor.
Q: In terms of product and systems development, what do you think will remain the same over the next decade? What will change?
Steven Meadows, Jama Software:
A lot of the areas I mentioned that will trend in 2022, will more than likely trend over the next decade.
AI, not only in medical but across most industries, is on the rise and patients will continue to see improved outcomes, quicker diagnoses, and a better quality of life.
Medical wearable technology will continue to evolve, with improved functionality helping keep us fitter and safer.
Minimally invasive devices will continue to be expanded across different medical areas, improving recovery times, and surgery outcomes. Increasing emphasis on cybersecurity will resume, to prevent malicious actors from hacking sensitive data and for connected systems to remain operational.
COVID-19 is not going away any time soon so a reliance on collaborative product development tools, like Jama Software, will continue to be an integral part of any organization placing an importance on quality and overall product and patient outcomes.
Ryan Moore, Jama Software:
Medical devices will still be comprised of mainly hardware (HW)/software (SW), while automation and robotics will be a driving force.And users will still need to power devices.
Carleda Wade, Jama Software:
Medical device development will mostly stay the same as the regulations are fairly stable.However, I do expect increased usage of software in the process since much of the workforce may now be working in various locations. I also expect to see an increase in AI and Software as a Medical Device (SaMD)as a whole in the industry.
Q: What are some of the biggest challenges you think engineering firms will be working to overcome in 2022?
Thierry Marchal, Ansys:
Contrary to other industries, innovation in healthcare faces the challenge of human variability: as we are all different, it is not acceptable that a treatment would work well for a few people and poorly for most others. Using computer modeling and simulation (CM&S a.k.a. in silico methods) is a cost-effective way to test new treatments efficiently without compromising with patient safety, on large cohorts of virtual patients.
An accurate prediction of the treatment outcome for a given patient will require combining traditional modeling techniques with biological models, often extracted from big data observations using AI.
Finally, the community remains skeptical about the reliability of computer model and digital evidence to predict correctly and accurately what is happening in the real life. This credibility challenge will be continuously addressed by more validations and comparisons with in vitro and in vivo data.
Ivan Ma, Experienced Medical Device Developer:
The development of medical device hardware will always require teams to gather in front of and handle hardware. As the pandemic continues, and in-person work still not back to what it once was, it is time to think of strategies that maximize time on developing hardware with minimal people in the room. Could digital Operating Room technologies be reconfigured and brought in to support the verification and validation phases of medical device development? The tenets are the same. Minimize the number of people in the room, allow the expert to drive the hardware from afar, make data acquisition, observation and learning access as easy as clicking on a link.
Q: How do you foresee regulations shifting in medical device product and systems development over the next decade?
Steven Meadows, Jama Software:
Although AI has been utilized in medical device and life science products for decades, guidance has been lagging. It’s clear to see that AI has incredible benefits for patients, and so there will continue to be increased regulatory guidance available, to help developers build products which contain AI in a safer and standardized manner. Check out a blog Jama Software authored around machine learning in SaMD and shifting regulations here.
The medical device regulation (MDR), which was accepted and implemented in the EU in 2017, has been amended over the past few years. The latest change focuses on increasing the responsibility and accountability of medical device companies throughout the entire development of a product. Expect more guidance over the next decade, as gaps are addressed to ensure product safety is at the forefront of any market clearance.
Ryan Moore, Jama Software:
I would expect compliance and regulations to be more defined as we move forward. Currently, there is a lot of grey area in how standards/regulations are interpreted. The FDA will need clearer guidance for modern technology that arises
Carleda Wade, Jama Software:
With an uptick in AI and SaMD I expect the FDA and foreign regulatory bodies to begin providing more clarity on how these complex systems and softwareneeds to be developed, validated, and maintained.
Q: What changing regulatory guidelines do you anticipate having an impact on companies in 2022?
Thierry Marchal, Ansys:
The US authorities developed, without any doubt, the most advanced regulation in terms of adoption of computer model results for the regulatory approval process. However, the number of published cases reporting the actual use of in silico methods by sponsors remains limited. New results will be published in 2022 further encouraging companies to confidently follow this process.
The European Medical Device Regulation (MDR), in application in Europe since May 2021, is opening the door to digital evidence and in silico approach; unfortunately, the process to validate a model and report simulation results has not been clarified yet. Similarly, as the European authorities are updating their pharmaceutical strategy in 2022, this document is expected to make references to computer modeling and simulation.
In the rest of the world, we observe a growing interest for in silico methods and the need to regulate this approach. After years of compiling information and experience from other parts of the world, we are expecting that some Asian authorities will start to communicate about this topic in 2022.
Q: What sorts of process adjustments do you think medical device development teams will need to make to be successful in 2022?
Steven Meadows, Jama Software:
One of the biggest process issues we see across our customers is that they view quality as a secondary function and a necessary checkbox activity once a product is developed, or close to being finalized. Because of that, products tend to contain more defects, resulting in more in field CAPAs, negative patient outcomes and even have a greater chance of being recalled. Development teams should ensure quality is prioritized from the get-go.
Although we have noticed a large shift with software teams adopting an Agile approach when it comes to development, we can’t emphasize enough the importance of adopting a lean and iterative approach.
Ryan Moore, Jama Software:
Continue to follow standardized process with focus on aligning with FDA / compliance in parallel with building safe products
Carleda Wade, Jama Software:
Teams will need to find the sweet spot of trying to collaborate and innovate while not being physically in the same location at the same time. Due to the pandemic, many teams will now transition to being fully remote permanently or have a hybrid schedule and it may make things more difficult when developing new products with a cross-functional team.
Q: From an engineering toolset perspective, what are some of the processes you believe forward-thinking firms will be working to leverage or incorporate into their process and why?
Theirry Marchal, Ansys:
As we see the healthcare community adopting in silico method with more enthusiasm both for design and regulatory approval (not mentioning emerging clinical applications), the software community is rushing to deliver the supporting in silico tools. We could mention three major avenues followed by Ansys.
As in silico clinical trials requires a large number of simulations, specific Simulation Process and Data Management (SPDM) tools structuring the digital evidence following the ASME VVUQ40 standard will greatly facilitate the adoption of this approach. Ansys is customizing its Minerva tool in a Minerva VVUQ 40 template for medical device and pharmaceutical companies.
It is crucial to model the behavior of a new treatment in its working environment, usually the human body. Human organs such as the heart, the lungs, and the brain are extremely complex to model and validate: this requires a collaboration of various actors. Ansys is closely working with leading academics, global healthcare companies and startups, and clinicians to continuously update and validate virtual hearts, lungs, and brains in collaboration with a large ecosystem.
Although everybody recognizes the potential of digital twin and Personal Digital Avatar, the necessity of developing quasi-instantaneous modeling capabilities despite the complexity of the model is a real challenge. Ansys is taking advantage of its experience with other industries already using digital twin of equipment to help the pharmaceutical industry to develop digital twin of drug manufacturing equipment and initiate the first steps towards Personal Digital Avatars.
Ivan Ma, Medical Device Program Lead:
Engineers need tools that minimize the volatility and error that occurs when bringing hardware and electronics from the drawing board to parts in hand. How do organizations reduce hardware prototype cycles, recognizing that even if the design is right on paper, the production of that design can pass through many hands before it is made into a real part, thus decreasing the probability of a successful build.
Q: What do you think will be some of the differentiators between a company surviving to see 2030, and those that do not?
Steven Meadows, Jama Software:
Along the lines of what I mentioned companies should focus on in 2022 to be successful, medical device companies that embrace a proactive approach to quality(Link here – https://www.jamasoftware.com/blog/three-ways-to-proactively-vs-reactively-incorporate-design-controls-in-medical-device-product-development/ )will ultimately find fewer issues with their products, improve customer satisfaction, and stay competitive for the foreseeable future. Companies that invest in best-of-breed medical device development solutions like Jama Connect will have the upper hand in reducing risk and complying with various standards and regulations.
New cutting-edge technologies in the medical field – like robotics nanotechnology, AI, and wearable health tech devices – bring complexities for medical device companies and risk for patients and consumers. Jama Software will continue to serve as the leading solution and partner to help innovative companies bring medical products to market, in a collaborative, safe, and efficient way.
Defined process, building quality products that meet a specific market need
Carleda Wade, Jama Software:
Truly understanding the market needs and being willing to allocate resources so that you become a pioneer in a certain industry will help a lot of companies to succeed; however, having passionate and qualified personnel will be the biggest success factor. Companies that only want to develop “me too” devices will struggle to gain market share, and even if they are able to survive it will be difficult for them to thrive.
Q: Where do you see Jama Software fitting in as the product development landscape evolves, and what can our customers expect as 2022 approaches?
Steven Meadows, Jama Software:
Jama Software provides incredible value for hundreds of medical device companies ranging from start-ups to established corporations. With COVID-19, we’ve seen an uptick in customers looking for an easy-to-use product development tool that offers differentiating collaborative capabilities to keep people connected throughout different design stages. Jama Software continues to invest heavily in the application, and we will see improved core product capabilities as well as integrations, strengthening the tool’s ability to seamlessly work alongside other systems.
Ryan Moore, Jama Software:
Jama Connect can be used as the core toolset for requirements, testing, and review/approvals. Jama Software’s ability to branch out into further workstreams (in order of priority: risk management, test automation, modeling/design outputs) will bring exponential value to teams using Jama Connect.
Carleda Wade, Jama Software:
I can see Jama Software bringing value to not only the start-up but the established company in the future. As a medical device company, the first goal of the company is to make a good product that meets a market need, this happens even before establishing a quality management system in many cases. Having a tool like Jama Connect will ensure that as products are developed that all the bases are covered. Later, when it is time for regulatory approval having a tool like Jama Connect in place will make the regulatory submission process very simple.
Thanks for tuning into our 2022 Predictions Series! To see some of the incredible products, software, and systems our customers are building with Jama Connect, visit our CUSTOMER STORIES PAGE.
https://www.jamasoftware.com/media/2022/01/2022-01-11_2022-Predictions_medical_1024x512-1-1.jpg5121024Decoteau Wilkerson/media/jama-logo-primary.svgDecoteau Wilkerson2022-01-11 03:00:352023-01-12 16:47:462022 Predictions for Medical Device Development: Increased Focus On Cybersecurity, Clarity On Standard Compliance, and the Challenge of Human Variability
Complying with FDA Design Control Requirements Using Requirements Management Principles
Mercedes Massana: So agenda for today is we’re going to talk a little bit about the design controls and what they are. We’re going to talk about requirements management and what that is. And then we’re going to talk about how the two relate to each other. Then we’ll discuss how we can build a good requirements management process and how that process can help us improve compliance to design controls.
Okay. So let’s get started. All right. So by the end of this presentation, hopefully you’ll have a good general working knowledge of design controls. You’re going to understand requirements management and how to evaluate and accept requirements, how to perform bi-directional traceability, how to use requirements attributes to help manage the requirements, and how requirements metrics can help you make better decisions. We’re going to learn how to manage and control requirements changes, and then how to use attributes, especially a requirements credit quality attribute to help us on manage and identify essential requirements and requirements that are critical to quality.
FDA Design Controls
Mercedes Massana: So let’s start with design control background. So design controls have been part of the FDA QSR since 1997. So it’s been a long time. The FDA initially implemented design controls to try to help medical device manufacturers identify deficiencies with design input requirements, to identify discrepancies between proposed designs and requirements, and increase the likelihood that the design transferred to production is going to translate into a device that is appropriate for the intended use and satisfies the user needs. And they did this in relation to having a lot of issues in the field with medical devices. So they thought design controls would improve the quality of the medical devices that were being approved.
In the FDA design control guidance, they state that developing a solid foundation of requirements is the single most important design control activities. So that kind of tells you how important FDA thinks design control requirements management is. So design controls is made up of several elements. There is design and development planning, design inputs, design outputs, design review, design verification, design validation, design transfer, design changes, and design history file.
Mercedes Massana: Now, most of those, even though requirements really falls under design inputs, and that’s what everybody understands by design inputs, most of these items relate to requirements in one way or another. So design outputs, we have to develop design outputs that will conform to design inputs. For design reviews, we’re going to review our design inputs to make sure that they’re appropriate and not conflicting or unambiguous. Design verification will confirm that design outputs meet the design input requirements. And design validation will confirm that the user needs and intended uses are met, which is their another form of design input.
And then design changes, obviously managing requirements changes is important. And obviously all our design inputs or all of our user needs documents or system requirements or product requirements documents will end up in the design history file. And then obviously our deliverables that user needs that we create, or system requirements, product requirements, all become part of our design history file.
Requirements Management
Mercedes Massana: So let’s talk about requirements management. So what is requirements? Why do we need requirements management? Why would we want to do this? Well, if your projects are late because you’re trying to introduce changes at the end, or if sometimes your developers can really figure out what the requirements may mean and they’re making their own interpretation, those are all issues that can be addressed through requirements management. In fact, it is said that approximately for IT projects, which encompass software obviously, that at least 50% of projects are late, or delivered with reduced functionality or over budget.
Mercedes Massana: If you have a good requirements management process, this will improve those statistics and make projects more successful. So what is a requirement? So for IEEE, a requirement is a property that a product must have to provide value to a stakeholder. That’s the IEEE definition. The FDA definition is a condition or capability needed by a user to solve a problem or achieve an objective. So couple of key items here is, something that is needed by somebody, right? So that tells you that anything that goes into your requirements has to have a purpose. Why is it there? And it’s needed to solve a problem. So it has to solve a problem for the user, right? So if it’s something that a software developer thinks is a cool feature, but nobody needs it, then it really shouldn’t be a requirement.
So requirements management process has four main goals. It tries to ensure that there is an understanding of requirements, but all the stakeholders, that they commit to those requirements or approve or agree that those are the requirements that need to be implemented. That we manage requirements changes. And we’ll talk what managing requirements changes means a little later. And then we identify inconsistencies between project work and requirements. And what that means is that you can’t have a project plan that says you’re going to be done in a year if you have 10,000 requirements to implement, right? So you have to have consistency between the requirements and the other deliverables of the project.
So to obtain an understanding of the requirements, first, we need to know who are we solving the problem for? Right? So we need to determine how we identify the stakeholders. So who are those requirements providers, right? Those are the stakeholders. We need to know how we’re going to evaluate the requirements, how we’re going to know if the requirements are good and complete. And then we need to know who needs to approve the requirements and when do requirements get approved. So that’s how we obtain an understanding of requirements.
Then a commitment to requirements is basically that agreement between the different stakeholders that says, if you develop these set of requirements, we’re going to have satisfied customers, that our agreement is usually made through approval of the requirements.
Managing requirements changes is, obviously the one constant in life is change. So requirements will also change. But it’s very important to manage changes so that the rest of the product reflects the impact of those changes, right? Nothing comes for free, and requirements changes usually are either going to cost more, and we’re going to have to add more resources or we’re going to have to add more scheduled, but something will have to be done in order to manage the impact of that change.
Requirements Traceability
Mercedes Massana: And then maintaining bidirectional traceability. This requires that requirements be traced forward from higher level requirements to lower level requirements and that every lower level requirement be able to be traced backwards to its parent or a higher level requirement. Additionally, other elements like design, tests and risks should also be included in our traceability. And later on, I’ll show you a fully elaborated trace matrix that shows the relationship between pretty much every deliverable that we’ll create as part of our development process.
All right. So what’s the connection between requirements management and design controls? So this table will show us a little bit of that. So these requirements management elements relate to design controls as follows. So design inputs are defined as the physical and performance requirements of the device that are used as the basis for device design. So the FDA tells us that design inputs must be appropriate. That we need to address incomplete, ambiguous or conflicting requirements. And that these requirements related to design inputs are addressed by the requirements management element and obtaining an understanding of requirements.
Design outputs are defined as the results of the design effort at each design phase and at the end of the total design effort. The FDA says that design controls must be able to evaluate the design outputs conformed to design inputs, that we must be able to establish acceptance criteria so that we can verify the design outputs conform to the design inputs. And then we identify the essential requirements or essential to proper function requirements. So again, requirements management can help us meet this part of the regulation.
FDA design controls also state that we must identify document, verify, validate and approve design changes before implementation. Additionally, the design control guidance specifies that the trace matrix is a form of verification. So both of these areas are also covered by requirements management. And here, we can see that there is definitely a relationship between FDA design controls and any requirements management process.
https://www.jamasoftware.com/media/2021/11/WB-2021-10-20_Complying-with-FDA-Design-Control-Requirements_Social-Image-1.png5121024Jama Software/media/jama-logo-primary.svgJama Software2021-11-11 03:00:122023-01-12 16:47:55Complying with FDA Design Control Requirements Using Requirements Management Principles
Over the last decade, digital health solutions have become increasingly important, powering revolutionary developments in areas such as robotics, genomics, diagnostics, patient/provider interactions, and many more.
In our recent webinar with MDM Engineering Consultants and BeanStock Ventures, we talked about critical standards and best practices to follow. Attendees also heard critical insight from industry leaders in digital health development.
Watch the webinar or read the recording to learn more about:
Digital health solution classifications
The most common digital health development challenges
How Jama Connect can be leveraged in your development process to help ensure FDA compliance
Industry leading insight and knowledge
Below is an abbreviated transcript and a recording of our webinar, Ensuring FDA Compliance for Your Digital Health Solution.
Mercedes Massana: Digital health merges different digital technologies to make health and healthcare applicable to our living in society and make it more personalized for the patient. Many people see digital health as the future of healthcare. Some things that are digital health are like wearable devices. For example, your Apple Watch. It has some sensors that can do an EKG. That would be considered a wearable device. Things like telehealth and telemedicine, which we all became more familiar with because of COVID, are also part of digital health.
For digital health to use technologies such as different computing platforms connectivity, which is very important because you’re trying to get information to the healthcare provider, software plays a huge and significant role in digital health, and then different technology with sensors. So these technologies are intended to be used as a medical product or as a companion diagnostic to a medical product and help the healthcare provider make decisions. Software is playing an increasingly important role in healthcare in general. As medical devices have gotten more complex, software has played a bigger role.
Three Types of Software in Medical Devices
Mercedes Massana: There are three different types of software related to medical device. One is software in a medical device, embedded in a medical device. Another is software as a medical device, which we’ll call SaMD, and we’ll be discussing today. And the third is software used to produce or maintain a medical device. All these three types of software are regulated by the different regulatory agencies. Software as a medical device, is software that is intended to be used for one or more medical purposes that perform these purposes without being embedded in a medical device. So when we say embedded in a medical device, it’s software that’s physically part of a medical device. Like an infusion pump has embedded software in it that runs on the GUI on infusion pump.
This means that this software that’s a medical device in itself is not something that’s tangible. It’s not something that you can touch, like a medical device where you can touch. Well, this software lives on the ether and it’s not something you can touch. But it provides meaningful information that can help you mitigate disease, it can help you diagnose screening. For example, if you had software that took information from an MRI and helped define whether cancer was present, that would be software as a medical device. It takes information from another device and it helps a healthcare provider and make a diagnosis.
Determining the Intended Use of Software in a Medical Device
Mercedes Massana: So for software as a medical device, an important part is to determine what that intended use is. And in order to do that, guidance documents provide… You have to provide the answers to these three very direct questions. One is what is the significance of the information provided by SaMD to the healthcare decision? Is this information going to be used to help treat a disease, to help diagnose a disease, to help clinical management of the disease like diabetes, or to just inform clinical management? Obviously, it’s more critical if it’s going to treat or diagnose a disease. We also want to know the healthcare situation or condition, the scenario in which the software is going to be used and whether absence for incorrect information at that time can result in critical and putting somebody in a critical position, where they might be subjected to serious injury. Or in a serious position where they might be subjected to additional procedures and things that would not be necessary. Or in a non-serious position where it’s just information is not really that critical.
And then we also want to know what is the core functionality of the software. What are the what is it supposed to do? What are his critical features and functions? So based on these three questions, we would craft an intended use statement for the software as a medical device. The healthcare situation, there’s three different classifications, as we said. Critical is where accurate or timely diagnosis or treatment action is vital to avoid death or serious injury. Serious is where we need accurate and timely information in order to avoid an unnecessary intervention. And then non-serious is where the information is important, but it’s not critical in order to avoid that serious injury or unnecessary intervention.
Using Categories provided by International Medical Device Regulators Forum (IMDRF)
Mercedes Massana: Once we’ve determined what that healthcare situation or condition is, then we can determine the significance of the software. We use the categories provided by IMDRF, which is the International Medical Device Regulators Forum. This organization has actually generated some guidance for SaMD. And it’s used by regulators to try to have some common level of standards on how the software is going to be regulated. So in order to get to the classification, we determine the healthcare condition, what’s that healthcare scenario. And then we determine what that information provided is going to be used for. So if it’s going to be used to treat or diagnose disease, and it’s in a critical scenario, that will have the highest level of criticality, which is a four. There’s four levels of classification one being the least critical.
Again, it’s a level of one to four. Four has a very high impact and one in low impact. So, a device that would screen for cancer or something that software would be used to screen for cancer, would be very high impact. Something where maybe we’re sending a heart rate for somebody who’s going through cardiac rehabilitation, that might be a lower impact because it’s just using to inform the clinician on how things are going, but it’s not being used directly to impact on the treatment. There are additional ways to classify medical devices. The global harmonization task force has a classification of A, B, C, or D. So four stage classification, where A is the lowest level of risk, and D is the highest level of risk. These can be used because SaMD is in itself a medical device, we can use this level too.
Mercedes Massana: Additionally, IEC 62304 classifies software in three different classes, C, B or A, where C is if the software fails, it can result in serious injury or death. B it can result in non-serious injury. And A would result in no injury or harm. Now, why is it important to classify software? Because this will tell us how much effort we need to put into the documentation and testing and so forth of software. Obviously, the more critical it is, the more effort it will be required. FDA also classifies software. They have a level of concern for software. And there’s three levels also which align pretty well with the definition of IEC 62304. For FDA, they use major, moderate and minor.
Software in general has always been challenging for manufacturing. Software as a Medical Device (SaMD) poses some additional challenges to the developer of the software. Some of these are, for example, the use of off-the-shelf software is a lot more prevalent in SaMD than it is with embedded devices. Regulatory agencies want to see a lot of information when you’re using off-the-shelf software or suit, which is software of unknown providence, so there’s a lot of requirements that you need to meet. This is essentially software that you don’t own, you don’t have the source code for, but it’s still used in your medical device, so you’re still responsible to ensure that it’s safe within the parameters that you’re using it. Software as a Medical Device also, it will run on different platforms. It can run on a computer. It can run on an iPad, it can run on a smartphone as opposed to embedded software where you know the environment that it will always operate in. So it is a challenge to ensure that software behaves effectively in any of the platforms that it will run it.
Challenge: Interconnectivity with Other Systems
Mercedes Massana: Even though most medical devices today have some form of interconnection right to other systems, for SaMD, it’s almost given that it will have to be interconnected because the information will have to get to the healthcare provider, and that is typically through the internet. It’s going to be sent… There will be connectivity to other systems that will provide challenges like cybersecurity. We need to keep information safe. Those are challenges that we need to meet. Secondly, the method of distribution, the frequency of updates of SaMD is different than that of an embedded medical device. So for an embedded medical device, the device gets built, the software gets put in EPROMS that gets built into the device, the device gets packaged. It gets sent out to a pharmacy where you might buy it or it gets sent out to a hospital where it will be unpacked and tested.
Well, Software as a Medical Device is not a tangible thing, so it’s going to be distributed through the internet there is no checks and balances there. There’s no checksum to know that you’ve downloaded it correctly, so it proves some additional challenges there. And then lastly here, it’s much easier to duplicate software that’s on the internet that you can download. The manufacturer doesn’t always have control of how this software is duplicated, as opposed to software for an embedded medical device where in the manufacturing facility it gets thrown into EPROMS and then gets put into the medical device. So the manufacturer has control. For SaMD, the manufacturer does not have control, or does not always have control.
Mercedes Massana: For most applications, the apps we have on our smartphones, they get updated probably three, four, five, six times a year. So for a medical device software, you need to have a pretty rapid development time in order to keep up with this change of information. Then there’s also cybersecurity risks that we need to be aware of. Again, information security is a big issue. SaMD is something that regulatory agencies are very interested in. How do we get to meet these development challenges? Well, we need to have a very robust design development process to help us meet these challenges. We need a process that’s very methodical and systematic. We need a good quality management system. And then we need a software lifecycle process that takes us through the entire software lifecycle for the development of this application. It should take us from the point of where we’re starting to understand user needs, all the way to where we’re ready to retire the software. IEC 62304 is such a process where most people that are developing medical device software are utilizing this process.
Challenge: Managing Software Changes
Mercedes Massana: In addition, because the environment that the software is used and can change after release of the software, it is important to have a very strong post-market surveillance process to monitor customer issues in the field. You also need to monitor for cyber security threats, things like that. And you might need to make changes to your software based on what you’re observing in the field. So a post-market surveillance process is very important. Additionally, managing software changes, which is always important no matter what the software is used for, whether it’s a Software as a Medical Device, or embedded software in a medical device, is very important to manage changes. But for Software as a Medical Device, it’s even more important because what you change in the software could actually change the classification of the software. It could be more critical or less critical and it can also change the intended use. If you change the intended use, that means that you’re going to have to submit to regulatory agencies, so you want before you get approval to market that software.
There are a lot of software applications. Now, Software as a Medical Devices where they’re getting so much data and information that they’re using artificial intelligence, or machine learning to make the software smarter and better. Using this could potentially change the classification of the software. SaMD is still fairly new to regulatory agencies so I don’t think they’ve figured out exactly how they’re going to regulate things like artificial intelligence and machine language. But managing changes and understanding the impact of those changes is very important for our Software as a Medical Device. There aren’t a lot of guidance documents out there for Software as a Medical Device, but luckily there are a few standards that can guide us in our development efforts.
https://www.jamasoftware.com/media/2021/09/2021-08-19-digital-health-webinar-1024x512-1.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2021-09-07 03:00:502023-01-12 16:48:06Ensuring FDA Compliance for Your Digital Health Solution